
咖啡因分子印迹聚合物微球的悬浮聚合法制备及吸附性能研究
2015-04-25 08:13:00李兆峰
三明学院学报 2015年4期
董 雁,李兆峰
(龙岩学院 化学与材料学院,福建 龙岩 364012)
分子印迹技术(MIT)基本原理是在一定的溶剂中加入印迹分子和功能单体,两者之间形成复合物,然后加入交联剂使其形成分子印迹聚合物(MIP),最后洗脱掉印迹分子。自二十世纪80年代提出分子印迹技术与分子印迹聚合物的概念后,这项技术逐渐被人们熟知。由于分子印迹技术可以有效地对于目标分子进行识别、检测、分析、分离等工作[1-4]。因此该技术具有很宽的应用范围,在环境、医药、食品、军事等方面,都有良好的应用前景。
咖啡因(化学名1,3,7-三甲基黄嘌呤或3,7-二氢-1,3,7三甲基-1H-嘌呤-2,6-二酮)是一种生物碱,从茶叶、咖啡果中提炼出来的,适量使用具有祛除疲劳、使神经兴奋的作用,临床上用于治疗昏迷复苏和神经衰弱。但是,大剂量或长期使用也会对人体造成损害,且有成瘾性,因此被列入受国家管制的精神药品范围。建立一种快速高效测定咖啡因含量检测方法具有重要意义。制备咖啡因及其类似结构印迹微球聚合物已经有报道,但多数采用沉淀聚合法[5-8]。本文采用悬浮聚合法在水环境中制备对咖啡因具有识别功能的印迹聚合物微球。
1 实验
1.1 主要仪器和试剂
1.1.1 仪器
TU-1901型双光束紫外可见分光光度计 (北京普析通用仪器有限责任公司);S-3400N钨灯丝扫描电子显微镜(日本日立公司);IS10傅立叶红外光谱仪(Thermo Fisher Scientific);STA-449F3同步热分析仪(德国耐驰公司)。
1.1.2 试剂
咖啡因(CP);聚乙烯醇(AR,PVA);α-甲基丙烯酸(AR,MAA);乙二醇二甲基丙烯酸酯(AR,EDGMA);偶氮二异丁腈(AR,AIBN);丙烯酰胺(AR,MA);三氯甲烷(AR)。
1.2 实验方法
1.2.1 印迹聚合物微球的制备
咖啡因分子印迹微球的制备:称取一定量的PVA,在90~95℃的温度下将其溶解到150 mL蒸馏水中,倒入150 mL三口烧瓶中冷却至室温后按一定的配方加入咖啡因三氯甲烷溶液、功能单体、交联剂EDGMA、引发剂AIBN,然后在室温下超声脱气30 min,在氮气保护下60℃反应24 h,所得印迹聚合物微球用蒸馏水洗涤至紫外可见分光光度计检测不出模板分子为止,将所得聚合物微球抽滤、干燥保存备用。非印迹聚合物(NIP)的制备除不加模板分子,其余步骤同上。配方和用量见表1~表 4。
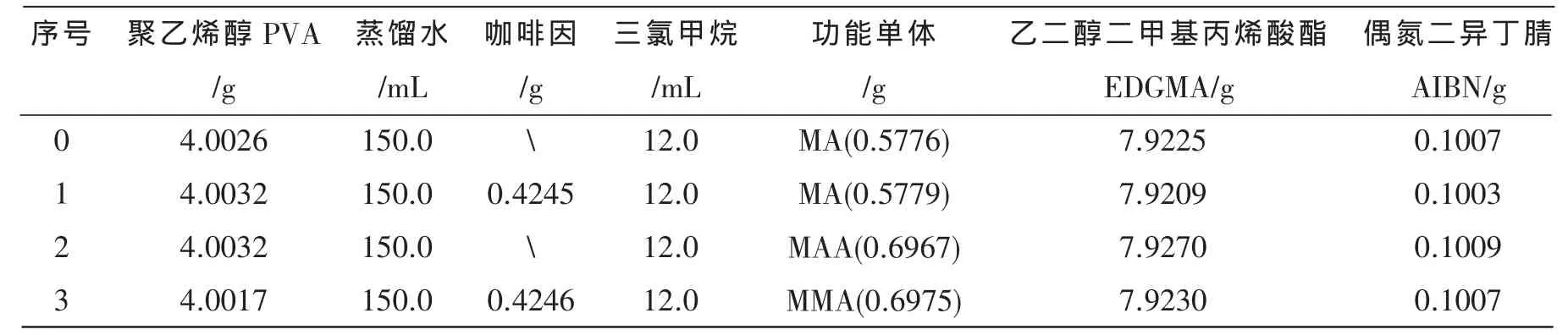
表1 不同功能单体的聚合物的配方和用量

表2 不同的模板分子与功能单体比例的聚合物的配方和用量

表3 不同的模板分子与交联剂比例的聚合物的配方和用量

表4 不同反应时间的聚合物的配方和用量
1.2.2 印迹聚合物微球的吸附性能的测定
配置浓度为0.5 g/L的咖啡因标准水溶液,取25 mL标准水溶液至于标有0到21号的25 mL容量瓶中,称取干燥的各组印记聚合物微球0.1 g加入到对应标号的容量瓶中,振荡放置24 h,然后用紫外可见分光光度计在272 nm下测定吸附后溶液的吸光度值,根据标准溶液中咖啡因浓度的变化得出印迹微球的相对吸附量Q。

式中C0为咖啡因标准水溶液的初始浓度,单位为μg/L;C为吸附后母液中咖啡因的浓度,单位为μg/L;V0为咖啡因标准水溶液的体积,单位为L;m为参加吸附的聚合物的质量,单位为g。
用上述方法测定NIP的相对吸附量。
1.2.3 印迹聚合物的吸附速率
取若干份5.0 mL标准溶液,分别加入0.1 g聚合物后封口,每4h测一份,计算相对吸附量,平行测定3次取平均值。
1.2.4 印迹聚合物微球的扫描电镜分析
真空中用离子溅射器对微球样品进行喷金,后用SEM对聚合物微球进行扫描,观测微球的表观形貌。
2 结果与讨论
2.1 功能单体的选择
分别以MA、MAA为功能单体,在60℃下聚合反应所制得的微球的产量与吸附量如表5所示。
由表5可知,从聚合物产量考虑:以MA为功能单体时的产量较用MAA为功能单体时的产量低,可能是由于PVA和水与MA之间形成氢键的能力太强,降低了分散剂的作用,导致产量比较低;从吸附量考虑:以MAA为功能单体时所制得的聚合物微球的吸附量比较高,是因为MA与模板分子咖啡因形成的氢键作用比较强,不利于咖啡因的洗脱,从而影响了聚合物微球的吸附量。而不加模板分子咖啡因的空白聚合物对咖啡因也有一定的吸附作用,这可能是部分位于聚合物表面的单体与咖啡因产生了氢键作用的结果,属于非特异性吸附。因此后续实验选取MAA为功能单体。
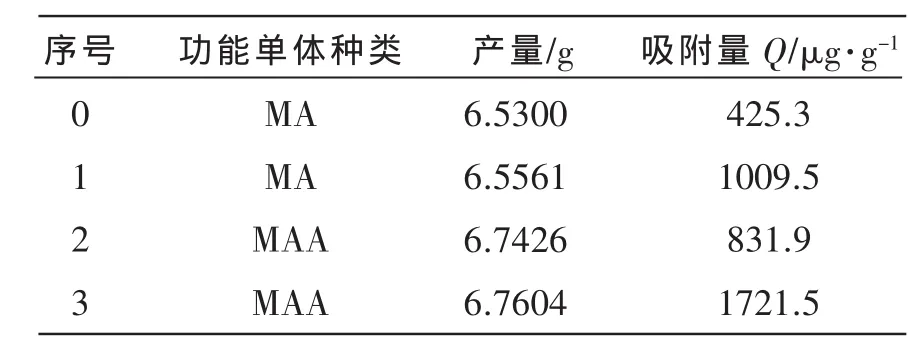
表5 印迹聚合物微球的产量及其吸附量
2.2 模板分子与功能单体的不同比例
在60℃下,不同的咖啡因与MAA的比例混合反应24 h,制得的印迹聚合物微球对咖啡因分子的吸附量Q,结果如表6所示。
从表6可知,随着功能单体用量的增加,吸附量呈现出由小变大再变小的趋势。这可能是因为单体用量较少时,咖啡因分子会两三个甚至更多个一组聚集在一起,在印迹聚合物微球表面形成较大的空穴,在与咖啡因分子识别过程中的结合作用微弱,不能有效的与咖啡因分子结合,影响聚合物的吸附量;当功能单体所占的比例增加时,吸附量也增大,当达到模板分子与功能单体间最佳比例n(咖啡因)∶n(MAA)为1∶4时,在该比例下,反应液中咖啡因分子恰好单个分子且能够比较密集的镶嵌在印迹聚合物微球表面并获取预定的取向和定位,在与咖啡因分子识别过程中表现出较强的结合力,能有效捕获咖啡因分子;吸附量也达到最大值,而随着功能单体比例继续增大,过量的MAA将部分咖啡因分子包在微球内部较难洗脱,聚合物微球的吸附量反而降低了[9]。

表6 不同模板分子与功能单体比例下印迹微球的相对吸附量
2.3 模板分子与交联剂的不同比例
60℃下,不同的咖啡因与EDGMA的比例混合反应24 h所制得的印迹聚合物微球的吸附量如表7所示。
从表7中可知,随着交联剂用量的增加,聚合物吸附量有着一个从小变大再变小的过程,在 n(咖啡因)∶n(EDGMA)=1∶20时,吸附量达到最大值。如果交联剂量太少,使得MIP不具有足够的交联度,形成的印迹“记忆”空穴结构不完善,分子印迹聚合物机械强度和化学稳定性较差,洗脱过程中模板分子留下的空穴容易被破坏,识别性能和结合强度下降,降低了MIP的吸附量;而交联剂如果太多,导致过交联度,使形成的印迹“记忆”空穴结构过于紧密,造成模板分子洗脱困难,MIP的吸附量也会降低。只有在最佳比例下,即模板分子与交联剂物质的量之比为1∶20,此时制得的印迹聚合物微球具有恰好足够的交联度,形成最适合的识别咖啡因分子的空穴结构以捕捉咖啡因分子,印迹聚合物微球具有最理想的吸附量。

表7 不同模板分子与交联剂比例下印迹微球的相对吸附量
2.4 反应时间
优化实验的反应时间(15-21号),找到一种最合适的反应时间,提高实验效率,计算在不同时间下制得的MIP的质量和吸附量,将不同反应时间条件下制得的MIP的质量和吸附量随时间的变化分别作图,如图1~图2所示。
由图1~图2可知,聚合物的产量和吸附量都随着聚合反应时间的增加而增大,但是在24 h时,产量和吸附量基本是达到了最大值,之后便不在随聚合反应时间的加长而增大。这可能是因为在反应时间较短时,聚合反应进程还在持续,随着微球数量在增多,微球表面还在继续形成有“记忆”的空穴,微球的产量和吸附量也就随着时间的加长而增多;当反应时间达到24 h时,基本完成了反应,微球表面的“记忆”空穴也不再增多,24 h后产量和吸附量保持稳定,而且产量和吸附量差不多是同步的增长。
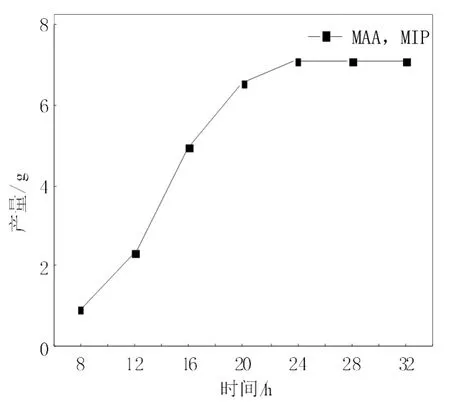
图1 不同反应时间条件下制得的MIP的产量随时间的变化关系
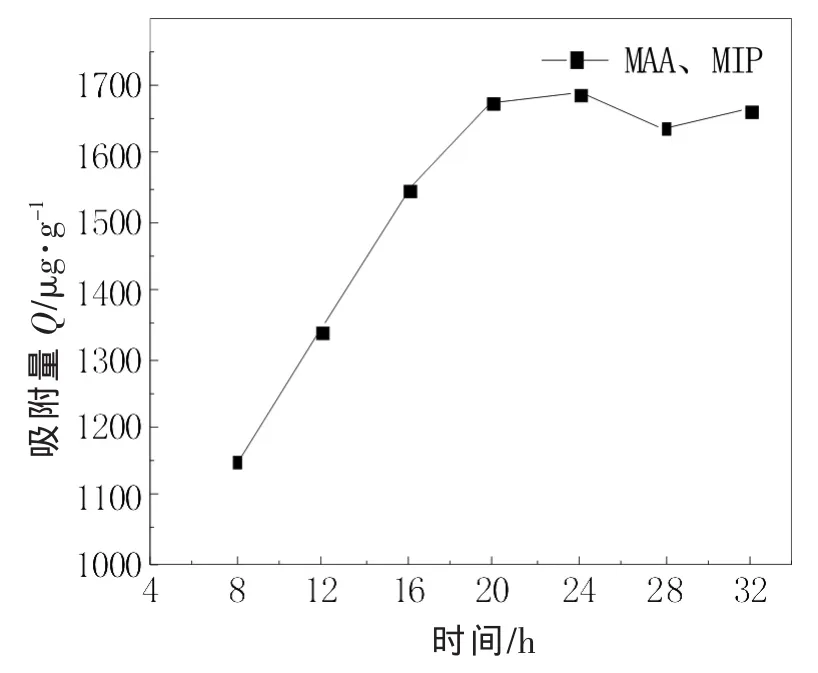
图2 不同反应时间条件下制得的MIP的吸附量随时间的变化关系
2.5 吸附速率的研究
测定所制备MIP在不同浓度咖啡因标准水溶液中吸附量随时间的变化曲线,如图3所示。
由图3可知,在吸附初始阶段浅孔对咖啡因分子的结合速率较快,但是,当浅孔被吸附饱和后,咖啡因分子向印迹微球的深孔传质有一定的位阻,导致结合速率下降,在20h内基本完成对咖啡因的吸附。
2.6 印迹微球的表观形貌
由图4可以看出,在放大500倍的下,聚合物呈现为微球形态,且粒径均匀。
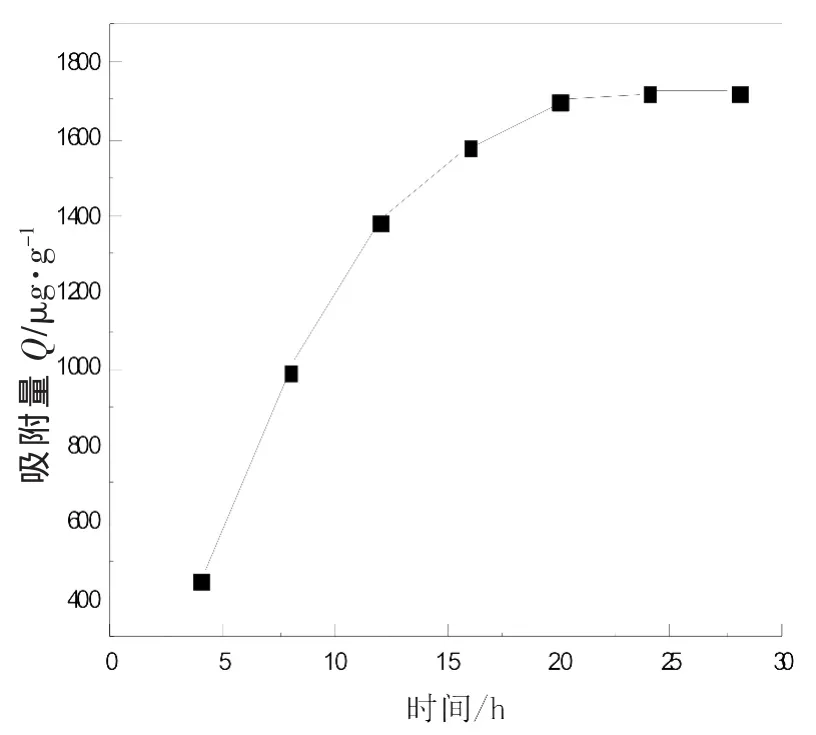
图3 MIP的吸附速率

图4 印迹聚合物微球扫描图
2.7 MIP的Scatchard分析
通过采用静态吸附法,测定以不同单体制成的印迹聚合物对咖啡因的结合等温线如图5所示。对获得的数据进行Scatchard分析用以评价聚合物的结合特性。Scatchard方程为
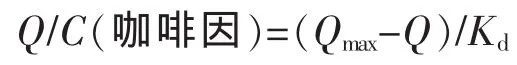
式中Kd(mmol/L)是结合位点的平衡离解常数;Qmax(μmol·g-1)是结合位点的最大表观结合量;C(咖啡因,μmol·L-1)表示聚合物达到平衡时上清液中的平衡浓度。以Q/C对Q作图,得到MIP的Scatchard曲线见图6。
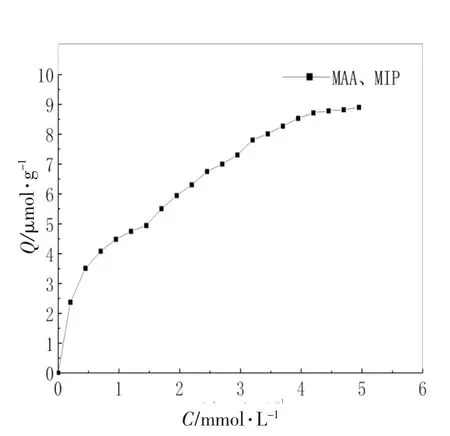
图5 MIP的结合等温线

图6 印迹聚合物的Scatchard曲线
由图 6 可知, 吸附量 Q 分别在 0~4.9423 μmol·g-1和在 4.9423~8.0991 μmol·g-1这两个范围内与Q/C呈线性关系,拟合可得到两个线性部分的拟合线性回归方程:y=19.47794-3.28676x,相关系数R=0.9985;y=5.29462-0.37737x,相关系数R=0.9869。由线性回归方程的斜率k和截距可以计算出高亲和位点的离解常数为Kd1=0.3043 mmol/L,最大表观结合量Qmax1=5.9262 μmol/g;低亲和位点的离解常数Kd2=2.6499 mmol/L,最大表观结合量Qmax2=14.0322 μmol/g。这表明在所研究的浓度范围内,印迹聚合物微球MIP对咖啡因存在有两类不等价的结合位点,具有非均一性[10],表明咖啡因分子印迹聚合物微球对咖啡因具有良好的再结合能力。可能是因为咖啡因分子结构中存在有酰胺基和氨基两种不同类型的功能基,均能与功能单体中的功能基(-NH,-OH)起作用,模板分子与功能单体预组装时存在着两种方式,可以形成两种不同组成的配合物,经交联后在聚合物中形成两种具有不同性质的作用位点。
3 结论
采用悬浮聚合法,在水溶液中合成对咖啡因具有选择吸附性的分子印迹聚合物微球,用紫外可见光分光光度计对印迹聚合物微球的吸附性能进行研究,结果表明,以α-甲基丙烯酸为功能单体合成的分子印迹聚合物微球的吸附性能较好,最大表观结合量可达14.0322 μmol/g。优化反应条件得出最佳的工艺条件为:模板分子、功能单体和交联剂的物质的量之比为1∶4∶20,反应时间为24 h。
[1]赖源发,张桂凤,游勇基.分子印迹技术原理及其在药物分析检测中的应用[J].海峡药学,2009,21(6): 4-8.
[2]GUAN G J,LIU B H,WANG Z Y ,et al.Imprinting of molecular recognition sites on nanostruc-tures and its applications in chemosensors [J].Sensors,2008,8(12): 8291-8320.
[3]LEONHARDT A,MOSBACH K.Enzyme-mimicking polymer exhibiting specific substrate binding and cataly-ticfunctions[J].React Polym,1987,6(3) : 285-290.
[4]MARRIA KEMPE,KLAUS MOSBACH.Separation of animo acids,peptides and proteins on molecularly imprinted stationary phases[J].J Chromatogrphy,1995,691 (2) : 317-323.
[5]梁哲,庄琼红,姚传义.沉淀聚合法制备1-苯丙醇印迹聚合物微球的研究[J].厦门大学学报:自然科学版,2007,46(2): 204-208.
[6]蒋红旭,吴嫦秋,刘展眉.茶碱分子印迹聚合物微球的合成及其性能研究[J].中草药, 2013,44(15): 2055-2058.
[7]王诚刚.咖啡因分子印迹的制备及其性能研究[J].广州化工,2009,37(8): 138-139.
[8]姚伟,高志贤,房彦军,等.沉淀聚合法制备咖啡因分子印迹聚合物微球[J].化工进展, 2007,26(6): 869-877.
[9]韩永萍,林强,王昶.悬浮法制备豆甾醇分子印迹聚合物微球[J].应用化学,2008,25(5): 556-559.
[10]李保利,张敏,姜萍,等.表面接枝分子印迹聚合物微球的合成及评价[J].化学学报, 2007,65(10): 955-961.
猜你喜欢
陶瓷研究(2022年3期)2022-08-19 07:15:18
云南画报(2021年10期)2021-11-24 01:06:56
粘接(2021年2期)2021-06-10 01:08:11
小学生优秀作文(高年级)(2018年4期)2018-09-11 01:23:22
大自然探索(2017年10期)2017-10-28 06:47:59
大自然探索(2017年5期)2017-05-26 17:48:07
实用临床医学(2016年8期)2016-06-07 01:28:16
石油化工(2015年9期)2015-08-15 00:43:05
橡塑技术与装备(2015年7期)2015-07-03 12:18:01
中国摄影(2014年12期)2015-01-27 13:57:04
