
原子分辨成像技术在锂离子电池电极材料中的应用
2015-04-23 10:57何敏肖东东谷林
新材料产业 2015年9期
何敏 肖东东 谷林
锂离子电池因其具有高的能量密度、高功率密度、长的循环寿命等优点而广泛应用于消费电子产品,如智能手机、笔记本电脑等。同时,一些新兴领域,如电动汽车和规模储能等,也表现出巨大的需求。如何进一步提高锂离子电池电化学性能以满足市场日益增长的储能需求已成为电化学储能领域面临的一个重要课题。
众所周知,锂离子电池主要由正极、负极、电解液和隔膜材料构成,其中电极材料是决定电池能量密度的关键因素。目前商用的锂离子电池主要是基于“摇椅式”的工作原理,充放电过程中通过往返于正负极之间锂离子可逆嵌入和脱出实现能量储存和利用。锂离子的充放电过程总是伴随着电极材料微观结构的显著变化,比如两相转变[1]、表面原子重构[2]、有序-无序转变[3]等,这都与电极材料的电压曲线、容量、倍率性能、循环寿命、储能机理等密切相关。因此,在原子尺度下研究电极材料在电化学循环过程中的结构变化,将有助于理解电极材料充放电机理及电化学演化规律,对于优化现有商业化材料体系和开发新材料具有重要意义。
透射电子显微镜(Transmission Electron Microscope,TEM)作为探索显微结构与性能关系的重要途径之一,因其高空间分辨特性已广泛应用于锂离子电池领域。但由于物镜像差(主要是受制于球差)的存在,在很长一段时间里,原子尺度结构信息都是通过对拍摄的高分辨图像进行复杂的图像处理获得。近些年,随着球差校正技术的实现,无论是传统的高分辨透射电子显微术(High Resolution Transmission Electron microscopy,HRTEM)还是扫描透射电子显微术(Scanning Transmission Electron Microscopy,STEM)都突破了1?的空间分辨率,使得直接获得原子尺度的结构信息成为现实。另外,基于球差校正TEM的谱学,如电子能量损失谱(Electron Energy Loss Spectroscopy,EELS)、X射线能谱(Energy-Dispersive X-Ray Spectroscopy,EDS)等,也同时实现了对材料的电子结构、元素组成和化学成分在亚埃尺度上的直接分析。然而,传统成像技术由于受限于轻元素相对较小的电子散射截面而几乎无法直接分辨出轻原子的空间位置,对电极材料关键的轻元素如锂(Li)、氧(O)直接成像仍然面临巨大的技术挑战。高角环形暗场扫描透射电子显微术(High Angle Annular Dark Field-STEM,HAADF-STEM)将汇聚到亚埃级别的电子束打在样品上并逐点扫描,通过置于样品下方的高角环形接收器接收电子成像。由于该成像技术图象衬度强烈依赖原子序数,近似与Z的1.7次方成正比[4],虽然可以获得可直观解释的原子分辨图像,但是无法直接观察轻元素。为解决轻元素观测难的问题,球差校正环形明场扫描透射电子显微成像技术(Annular Bright Field-STEM imaging,ABF-STEM)应运而生,其主要思路是弱化图像衬度对原子序数的依赖性,从而实现对轻、重原子的直接观察。该技术的出现,成功的实现了对轻元素,如Li、碳(C)、氮(N)、O、甚至氢(H)的直接成像。图1(a)为环行明场成像的光路示意图[5],成像过程与HAADF不同的是,置于样品下方的环形探测器收集的角度范围不同,导致所获得图像的衬度解释机理不同,ABF的图像衬度对原子序数的依赖被大大削弱,仅近似与Z的1/3次方成正比。还需要指出的是,不同于HAADF中亮点代表原子柱空间位置,在ABF中黑点标示原子柱位置。图1(b)、(c)分别为磷酸铁锂(LiFePO4)沿[010]方向的HAADF像和ABF像。可以看出,相对于HAADF像,ABF像在观察重元素的同时还可以分辨轻元素的空间位置。由于环形明场成像方法在原子尺度对轻元素直接成像的独特优势,目前已在电极材料表征方面获得广泛应用。
一、表面结构
众所周知,电池工作时在电极表面发生的电化学反应,如锂的吸附、电荷转移、固体电解质界面膜(SEI)的生成、电解制分解等,将不可避免地引起电极材料表面结构的变化,从而对电池性能(如倍率性能、安全性、循环寿命等)产生重要影响。因此,在原子尺度下阐明电化学循环过程中电极表面结构和化学成分的变化,不仅有利于更深入的认识电极材料的性能演化规律,而且为材料表面改性提供重要参考。
1.钛酸锂(Li4Ti5O12)中的表界面结构
Li4Ti5O12(LTO)是一种高功率、长寿命的负极材料,其优异的性能一方面是源于它嵌锂电位(1.55V vs. Li+/Li)高于商用石墨负极,可以避免在电化学循环过程中在电极表面形成SEI膜。另一方面,在充放电过程,Li4Ti5O12因具有在电化学循环过程中晶格常数基本保持不变的特性(在充放电过程只有0.2%的体积形变),也被称为“零应变”材料,具有非常好的循环性能。
立方晶系的Li4Ti5O12中,3/4的Li占据8a位置,O占据32e位置,剩下的Li和Ti占据16d位置。一般认为, Li4Ti5O12充放电过程是一个典型的两相反应,反应方程式可以表示为:Li4Ti5O12+Li++e-←→Li7Ti5O12。虽然对于两相反應机理目前已有比较深入的认识,但是仍然缺乏直观的微观结构细节。卢侠等[6]利用球差校正环形明场像技术首次直接观察到部分锂化后Li4Ti5O12与Li7Ti5O12的两相共存界面的原子结构细节,进一步证实了两相反应机制;并指出,不同于以往认为的核-壳模型的两相相对分布模式[7],Li7Ti5O12是由颗粒表面局部区域向内部延伸的。同时还发现,颗粒表面存在一层与体相不一样的结构,并认为可能与实际应用过程中的胀气有关[8]。卢侠等人[9]近一步研究发现,嵌锂后的Li4Ti5O12在表面形成了一层厚度约1.7nm的Li-钛(Ti)原子列重叠区域。如图2(a)所示,这是由于表面晶格膨胀诱导了16d位置的Ti离子迁移到16c锂离子晶格位置处而形成与体相不一样的结构。为弄清楚表面处电荷分布,对该区域进行EELS线扫,线扫的区域为图b绿线处(需要指出的是为了获得更高的能量分辨率而相应的牺牲了空间分辨率,但是从图b中仍然可以看到基本的结构单元,b中的四边形)。一般认为,在电化学嵌锂过程中,LTO电极的反应过程可表示为Li++e-+Ti4+→Li++Ti3+,嵌入的锂离子和原晶格中处于8a位置的锂离子都迁移到16c位置,最终导致尖晶石结构Li4Ti5O12向盐岩结构Li7Ti5O12转变。然而,从图d数据表明相应的逆反应在表面处发生,即Li++Ti3+-→Li++e-+Ti4+。在电化学循环过程中伴随着该反应的进行,释放的电子将会影响电解质和SEI膜中有机物氧化还原过程。因此,自发的表面反应Ti3+-→e-+Ti4+在嵌锂的LTO胀气问题中扮演者关键的角色。
尖晶石结构的Li4Ti5O12虽然因其特有的电化学性质而被认为在长寿命锂电池中有巨大的应用空间,但是由于其本身低的锂离子扩散系数和比较差的电子电导,导致其高倍率性能较差,一般认为通过纳米化和表面包覆能有效解决这一问题。王永庆等[10]首次合成了二氧化钛(TiO2)侧面包覆的Li4Ti5O12纳米片,如图3所示,从a、b中的球差校正的HAADF像以及对应c中的ABF像可以看到TiO2包覆层沿Li4Ti5O12[001]方向外延生长,成功获得了很好的包覆层。电化学测试发现Li4Ti5O12的倍率性能得到明显的提高,这表明TiO2的包覆可以有效改善锂离子扩散速率和电荷转移反应。该研究结果进一步说明了表面改性对于优化现有电极材料性能的重要性。
2.锰酸锂(LiMn2O4)中的表界面结构

尖晶石结构的LiMn2O4最早是由M. M. Thackeray在1983年首次公开报道,由于具有高功率密度、高安全性能、低成本和环境友好等优点,目前已成功应用于电动汽车。然而,在电化学循环或长时间储存时LiMn2O4存在严重的容量衰退现象,限制了它的循环寿命。一般认为,容量的衰减与电极表面锰(Mn)的溶解密切相关,但是对于Mn溶解的机理,以及为什么Mn溶解的速率随Li的减少而增加等问题都需要进一步研究。由于Mn溶解与电极表面息息相关,因此理解这一现象的关键在于从原子尺度观察充放电过程中LiMn2O4表面结构变化。
唐代春等[11]利用球差校正HAADF首次在亚埃尺度下观察了LiMn2O4在电化学循环过程中的表面结构相变,详见图4。从图4(a)中可以看出当LiMn2O4脱锂时,在其表面处Mn原子从原先的16d位置迁移至原来Li所占据的8a位置,从而在表面形成了四氧化三锰(Mn3O4)相;(b)表明随着充电电压的升高,表面相转变区域进一步扩大,这意味着相变的程度与充电状态有关;(c)、(d)进一步证实表面相变对充电态的依赖性,随着放电过程的进行,当电压降到3.0V时,表面Mn3O4相逐渐减少并最终消失,恢复为与体相一致的相。因此,唐代春等认为表面Mn3O4的分解路径有2种可能性,一是Mn3O4中四面体位置的Mn2+溶于電解液中,恢复与体相类似的结构,二是表面形成的Mn3O4完全分解,溶于电解液中。不论是哪一种情况,都说明充电过程中表面形成的Mn3O4相是导致Mn溶解和活性材料损失的重要因素,通过有效的表面改性手段稳定表面晶体结构将可能是抑制Mn溶解的关键。
二、相界面
在众多电极材料中,存在一类电极在嵌/脱锂过程中发生两相反应,在电压曲线上表现为一个平坦的电压平台。例如,LiFePO4在脱锂时转变为磷酸铁(FePO4),Li4Ti5O12在嵌锂时转变为Li7Ti5O12。这类材料具有一个共同的特点,即电化学循环过程都伴随着两相界面迁移过程。这一过程决定了两相转变的动力学特性,并最终决定了材料的倍率性能。因此,理解两相反应机制首先需要解决的关键问题是如何获得两相界面的原子结构信息。本节以LiFePO4为例,总结最近对原子尺度界面结构的研究情况。
LiFePO4最早由美国德州大学奥斯汀分校的Goodenough等[12]在1997年首次公开报道,其后,大量研究工作集中在优化其电化学性能和阐释局部LiFePO4和FePO4的两相反应。经过多年不懈努力,LiFePO4已成功实现商业化。一般认为,LiFePO4在电压曲线表现的一个3.45V(vs Li+/ Li)电压平台,说明在充放电过程中经历一个两相反应,可以表示为: LiFePO4←→FePO4+Li++e-。多种模型被提出用于解释两相反应微观机理,例如马赛克模型(mosaic model)[13]、核-壳模型(core-shell model)[12]、多米诺模型(domino-cascade model)[14]、新核-壳模型(new core-shell model)[15]等。然而缺乏两相界面的结构细节,反应机制仍然存在很大争议。随后,Chen等[16]利用高分辨成像技术观察到介于FePO4和LiFePO4两相之间存在一个无序的界面区域。通过电子能量损失的进一步研究,Laffont等[15]认为这一区域的成分由FePO4和LiFePO4两相构成。由于当时的实验条件有限,以上研究并未给出相界面结构原子尺度的细节信息。
利用[17]利用ABF技术首次在LiFePO4中直接观察到对电极材料电化学循环过程起到关键作用的锂离子,并且第1次报道了半充电态的LiFePO4单晶纳米线中出现Li+隔行脱出的“阶”现象。如图5所示,从图5(a)中可以清楚的观察到初始样品LiFePO4中的Li+,在图中用黄圈标示出锂的空间位置,当完全充电后,Li+全部脱出,见图5(b),图中用橘色圈标示出Li+脱出后的空位位置。从图5(c)可以看出,当样品处于半充电态时,锂离子出现了隔行脱出的现象,这与石墨负极在充电过程出现的“二阶(staging-II)”单相结构类似。该发现意味着,不同于以往提出的两相反应模型,LiFePO4在脱锂过程中存在一个中间相。很快,Malik等[18]通过理论模拟计算表明LiFePO4在充放电过程中存在一个通过类似阶结构的中间相实现单相反应的可能路径。Orikasa等[19]和Liu等[20]分别利用原位X射线衍射和软X射线吸收光谱也给出了LiFePO4脱锂过程中存在单相结构的实验证据。这些新发现表明LiFePO4在非平衡态条件下(高倍率充放电)会通过中间相发生连续相变,这也从某种程度上说明了电子绝缘体的LiFePO4为什么仍能实现快速充放电。
为了阐明掺杂、颗粒尺寸等对阶结构的影响,Suo等[21]对Nb掺杂的LiFePO4纳米颗粒(d=200nm)在部分脱锂态的原子结构进行了研究,发现阶结构的出现不受Nb在Li位的掺杂的影响,这表明两相界面处的阶结构是本征的亚稳或中间相。此外,阶结构可能也提供了一种温和的锂离子输运通道,这意味的中间态的存在可能对电化学过程是有利的。如图6所示,Zhu等人[22]将LiFePO4颗粒减小到70nm时,观察到类似的阶结构界面,宽度增加到15nm。这种明显的LiFePO4/stage-II/FePO4三相共存界面与先前经典的两相模型不一致。研究表明,这种阶结构会受到颗粒尺寸的影响,但是这种阶是LiFePO4一种本征的结构,与样品的掺杂、形貌、嵌锂方法无关。毫无疑问,对阶结构的直接观察不仅对LiFePO4脱锂行为有
了新的认识,同时为理解充放电过程中相界面的迁移提供了重要的启示。为了进一步阐明二阶结构形成的内在机理,孙洋等[23]通过密度泛函理论计算研究发现,从热力学的角度而言,发生两相反应是有利的。而从动力学的角度而言,由于Fe2+/Fe3+氧化还原对与Li+相互作用使得锂离子隔行脱出更有利,在充电过程中出现LiFePO4/ Staging-II/FePO4三相共存结构是动力学和热力学影响因素的相互竞争的结果。由于理论计算并未考虑小尺寸效应的影响,这需要进一步研究的证实。
三、过渡金属离子迁移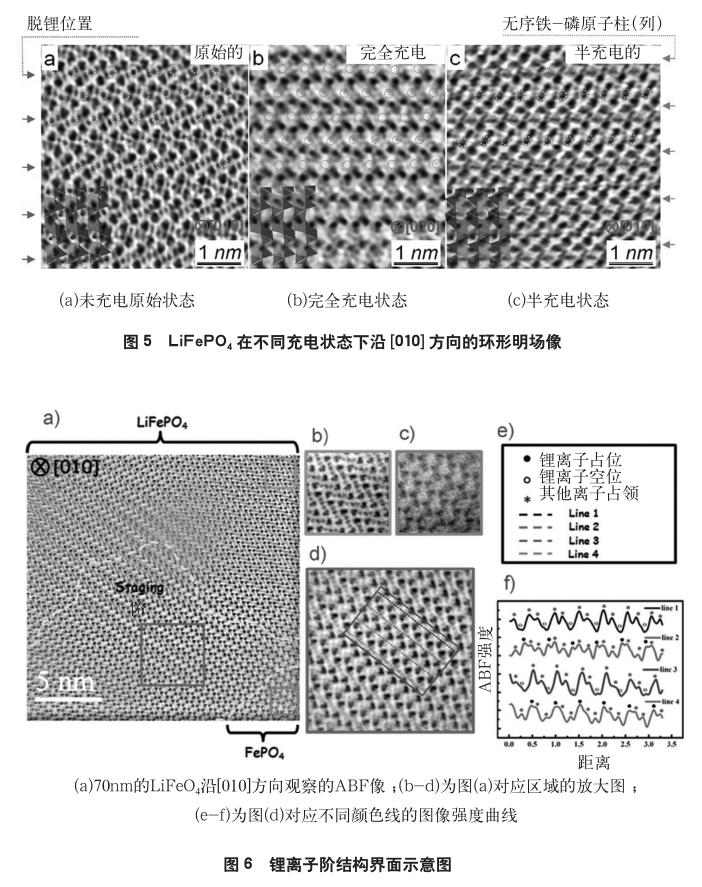
对于层状过渡金属氧化物电极材料而言,电化学性能好坏取决于材料原子结构和稳定性。为获得高的稳定性,可以通过在充放电过程中调控材料的骨架结构实现,而这往往取决于我们对骨架结构破坏机制的理解程度。在众多关于结构失稳的解释中,过渡金属离子迁移导致的阳离子混排(Cation Mixing)或者结构重排被认为是决定层状化合物结构稳定性的关键因素。富锂锰基层状氧化物正极材料(LMR)因其具有高电压和高比容量(大于250mAh/g)的特性而被看作是最有可能成为新一代商用锂离子二次电池正极材料之一。这类材料的通式可以表示为xLi2MnO3·(1-x) LiMO2,M代表过渡金属,如钴(Co)、镍(Ni)、Mn、铁(Fe)等。该类材料可看成由Li2MnO3和LiMO2按不同比例复合而成。其中,空间群为单斜的C2/ m的Li2MnO3中Mn層中有1/3的位置被Li+占据,形成有序的LiMn2层,因此Li2MnO3也可以表示为Li[Li1/3Mn2/3] O2。然而LMR存在的电压衰减、容量衰减和低倍率等问题阻碍了其商业化的步伐。该类材料在首次充电过程中,表现出与其组分材料Li2MnO3十分相似的电化学行为,体现在电压曲线上,是在4.5V左右存在一个明显的电压平台。因此,大量的研究工作试图通过理解富锂材料Li2MnO3在嵌/脱锂过程中的电化学性能和结构的关系,进而阐释富锂层状正极材料复杂的电化性能产生的机理,尤其是对电压衰减、容量衰减和低倍率性能机制的认识。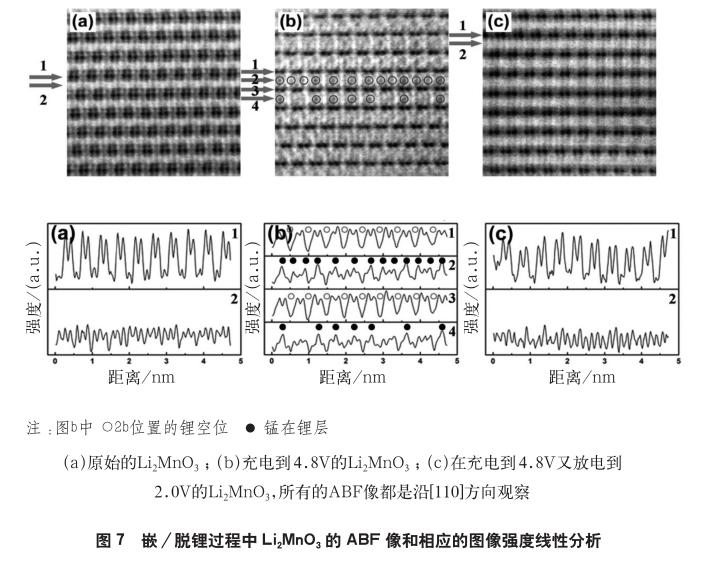
王锐等人[24]利用ABF技术研究了Li2MnO3在浅度(脱出的Li含量约为0.37)脱锂和嵌锂过程原子结构变化。研究发现,伴随Li+的脱出,Mn离子通过中间四面体迁移到锂层导致阳离子混排。如图7所示,黑色的点对应的原子列的位置。当沿着[110]方向观察Li2MnO3时可以看到,LiMn2层由2个相邻的Mn原子列(间隔0.14nm)和1个锂原子列交替排列而成,从图7(a)线1对应的像强度曲线可以清晰的观察到(波峰代表原子列位置)。经过初始的充电到4.8V后,LiMn2层的像强度曲线[图7(b)的线1和线3]只显示了2个相邻Mn原子列的重复信号,这表明LiMn2层的Li在充电过程可以和锂层的Li同时脱出。紧接着,在放电到2.0V时,从图7(c)线1处可以看出,之前脱出的Li离子又重新嵌入到LiMn2层。因此,LiMn2层的Li的脱/嵌过程是可逆的。另外,相比于原始材料的锂层[图7(a)线2],脱锂后的Li2MnO3的锂层[图7(b)线2和线4]出现一些灰点,在图中用圆圈标示,这意味着Mn离子在充电过程从过渡金属层迁移到了锂层。但是让人意想不到的是当放电到2V时,Mn又从锂层回到过渡金属层〔图7(c)〕,这意味着在浅充放电过程中,Mn的迁移过程也是可逆的。这与之前报道的Li2MnO3中Mn的不可逆迁移至Li层导致结构失稳不一致。对原子结构的直接观察表明Mn具有很高的移动性,但是对于产生这一现象的原因需要更深入的研究。目前有报道认为Mn的迁移会带来LMR层状正极材料内Li2MnO3成分的负面影响,导致LMR相对低的倍率性能[25]。此外,有研究表明Li2MnO3中Mn的动力学特性在LMR材料的电压滞后和电压衰减中起到了重要作用[26-29]。有模型认为电压滞后源于Mn的可逆迁移,而电压衰退源于Mn的不可逆迁移[30]。通过中子衍射,Mohanty等[31]证实Mn从过渡金属层八面体位置经过锂层四面体位置不可逆的迁移到锂层八面体位置从而导致向尖晶石结构转变,并最终导致LMR材料的电压衰减。通过原子分辨的HAADF技术,Sathiya等[32]指出,在富锂相中,阳离子在过渡金属层与锂层间的迁移是本征的属性。另外,电压衰退与在四面体位置捕获过渡金属离子密切相关。从该观点出发,这意味着由于结构重排导致的电压衰退不可能被完全消除。因此,在LMR材料电化学循环过程中如何最小化过渡金属离子迁移成为减缓电压衰减问题的关键。基于这种想法,Thackeray等[33-34]发现在xLi2MnO3·(1-x)LiMn0.5Ni0.5O2组分材料中,通过控制Mn和Ni之间复杂的相互作用可以有效抑制Mn离子的迁移,进而稳定结构,抑制电压衰减。该结论被Zhang等人[35]进一步证实,
該课题组发现通过在原子尺度提高元素分布的均一性从而增强Ni-Mn相互作用后,Li[Li0.2Ni0.2Mn0.6]O2正极材料的电压衰减得到了极大的改善。Ni稳定LMR材料结构的效应归功于其减少了放电过程中Mn4+向不稳定Mn3+的转变[36]。电极材料元素原子尺度间相互作用与材料功能性结构稳定之间的基本关联,为优化组分和减缓LMR材料的电压衰减提供了新的思路。
四、结语
电动汽车和规模储能市场迫切需要先进的表征手段揭示电池材料在电化学过程中的微观结构演化规律,以期提高锂离子电池性能,满足市场巨大需求。原子分辨的成像技术已使我们能够直接在原子尺度同时观察重元素和轻元素,为理解物质结构和性能的关系提供了重要机遇。本文简述了原子分辨成像技术如何揭示不同电化学状态下电极材料原子结构细节,成功地阐明了典型正负材料表/界面原子结构和电子结构、过渡族金属迁移。尽管如此,仍然有许多存在于材料制备和电化学循环过程的局部结构问题等待被解决,比如晶格畸变、空位、缺陷、断裂和表面腐蚀等。这些问题的解决将加深我们在原子尺度对电极材料Li存储、输运、相转变和结构衰退的理解,进而最大限度优化电极材料的电化学性能。
参考文献
[1] Yamada A,Koizumi H,Sonoyama N,et al.Phase change in LixFePO4[J].Electrochem Solid-State Lett.,2005(8):409-413.
[2] Xu Bo,Fell C R,Chi Miaofang,et al.Identifying surface structural changes in layered Li-excess nickel manganese oxides in high voltage lithium ion batteries:A joint experimental and theoretical study[J].Energy Environ Sci,2011(4):2223-2233.
[3] Shao-Horn Y,Levasseur S,Weill F,et al.Probing lithium and vacancy ordering in O3 layeredLixCoO2 (x approximate to 0.5)An electron diffraction study[J].J.Electrochem.Soc.,2003(150):366-373.
[4] Hillyard S,Silcox J.Detector geometry,thermal diffuse scattering and strain effects in ADF STEM imaging[J].Ultrami croscopy,1995,58(1):6-17.
[5] He Xiaoqing,Gu Lin,lu Xia,et al.Direct imaging of lithium ions using aberration-corrected annular-bright-field scanning transmission electron microscopy and associated contrast mechanisms[J].Mater Express,2011(1):43-50.
[6] Lu Xia,Zhao Liang,He Xiaoqing,et al.Lithium storage in Li4Ti5O12 spinel:the full static picture from electron microscopy[J].Adv Mater,2012(24):3233-3238.
[7] Amine K,Belharouak I,Liu Jun,et al.Electrochemical and thermal investigation of Li4/3Ti5/3O4 spinel[J].J.Electrochem. Soc.,2007(154):114-118.
[8] He Yanbing,Li Baohua,Liu Ming,et al.Gassing in Li4Ti5O12-based batteries and its remedy[J].Sci. Rep.,2012(2):1-9.
[9] Lu Xia,Gu Lin,Hu Yongsheng,et al.New insight into the atomic-scale bulk and surface structure evolution of Li4Ti5O12anode[J].Journal of the American Chemical Society.2015,137(4):1581-1587.
[10] Wang Yongqing,Gu Lin,Guo Yuguo,et al.Rutile-TiO2 nanocoating for a high-rate Li4Ti5O12 anode of a lithium-ion battery[J].J Am Chem Soc,2012,134(18):7874-7879.
[11] Tang Daichun,Sun Yang,Yang Zhenzhong,et al.Surface structure evolution of LiMn2O4cathode material upon charge/ discharge[J].Chem.Mater.,2014,26(11):3535-3543.
[12] Padhi A K,Nanjundaswamy K S,Goodenough J B.Phospho-olivines as positive-electrode materials for rechargeable lithium batteries[J].J Electrochem Soc,1997,144(4):1188-1194.
[13] Andersson A S,Thomas J O.The source of first-cycle capacity loss in LiFePO4[J].J Power Sources,2001,20(6):498-502.
[14] Delmas C,Maccario M,Croguennec L,et al.Lithium deintercalation in LiFePO4 nanoparticles via a domino-cascade model[J].Nat Mater,2008(7):665-671.
[15] Laffont L,Delacout C,Gibot P,et al.Study of the LiFePO4/FePO4 two-phase system by high-resolution electron energy loss spectroscopy[J].Chem Mater,2006(18):5520-5529.
[16] Chen Guoying,Song Xiangyun,Richardson T J.Electron microscopy study of the LiFePO4 to FePO4 phase transition[J]. Electrochem Solid-State Lett,2006,9(6):295-298.
[17] Gu Lin,Zhu Changbao,Li Hong,et al.Direct observation of lithium staging in partially delithiated LiFePO4 at atomic resolution[J].J Am Chem Soc,2011,133(13):4661-4663.
[18] Malik R,Zhou Fei,Ceder G.Kinetics of non-equilibrium lithium incorporation in LiFePO4[J].Nat.Mater,2011(10): 587-590.
[19] Orikasa Y,Maeda T,Koyama Y,et al.Direct observation of a metastable crystal phase of LixFePO4 under electrochemical phase transition[J].J Am Chem Soc,2013,135(15):5497-5500.
[20] Liu Xiaosong,Liu Jun,Qiao Ruimin,et al.Phase transformation and lithiation effect on electronic structure of LixFePO4:an in-depth study by soft X-ray and simulations[J].J Am Chem Soc,2012,134(33): 13708-13715.
[21] Suo Liumin,Han Wenze,Lu Xia,et al.Highly ordered staging structural interface between LiFePO4 and FePO4[J]. Phys Chem Chem Phys,2012(14):5365-5367.
[22] Zhu Changbao,Gu Lin,Suo Liumin,et al.Size-dependent staging and phase transition in LiFePO4/FePO4[J].Adv. Funct.Mater.,2014,24(3):312-318.
[23] Sun Yang,Lu Xia,Li Hong,et al.Kinetically controlled lithium-staging in delithiated LiFePO4 driven by the Fe center mediated interlayer Li-Li interactions[J].Chem Mater,2012,24(24):4693-4703.
[24] Wang Rui,He Xiaoqing,Wang Fangwei,et al.Atomic structure of Li2MnO3 after partial delithiation and relithiation[J].Adv.Energy Mater,2013,3(10):1358-1367.
[25] Yu Xiqian,Lyu Yingchun,Gu Lin,et al.Understanding the rate capability of high-energy-density Li-rich layered Li1.2Ni0.15Co0.1Mn0.55O2 cathode materials[J].Adv.Energy Mater,2014,160(5):3006-3019.
[26] Croy J R,Gallagher K G,Ren Yang,et al.Examining hysteresis in composite xLi2MnO3·(1-x)LiMO2 cathode structures[J].J.Phys.Chem.C,2013(117):6525-6536.
[27] Gallagher K G,Croy J R,Balasubramanian M,et al.Correlating hysteresis and voltage fade in lithium-and manganese-rich layered transition-metal oxide electrodes[J].Electrochem.Commun,2013(33):96-98.
[28] Bettge M,Li Yan,Zhu Ye,et al.Voltage fade of layered oxides:its measurement and impact on energy density[J].J. Electrochem.Soc.,2013,160(11):2046-2055.
[29] Croy J R,Gallagher K G,Balasubramanian M,et al.Quantifying hysteresis and voltage fade in xLi2MnO3center dot(1-x)LiMn0.5Ni0.5O2electrodes as a function of Li2MnO3content[J].J.Electrochem.Soc.,2014(161):318-325.
[30] Gallagher K G,Croy J R,Balasubramanian M,et al.Correlating hysteresis and voltage fade in lithium-and manganese-rich layered transition-metal oxide electrodes[J].Electrochem.Commun,2013(33):96-98.
[31] Mohanty D,Li Jie,Stan M,et al.Unraveling the voltage fade mechanism in high-energy-density lithium-ion batteries:origin of the tetrahedral cations for spinel conversion[J].Chem.Mater.,2014,5(4):6272-6280.
[32] Sathiya M,Abakumov A M,Foix D,et al.Origin of voltage decay in high-capacity layered oxide electrodes[J].Nat. Mater.,2015(14):230-238.
[33] Croy J R,Kim D H,Balasubramanian M,et al.Countering the voltage decay in high capacity xLi2MnO3 center dot(1-x)LiMO2 electrodes(M=Mn,Ni,Co)for Li+-ion batteries[J].J.Electrochem.Soc.,2012,159(6):781-790.
[34] Kim D H,Croy J R,Thackeray M M.Comments on stabilizing layered manganese oxide electrodes for Li batteries[J]. Electrochem.Commun.,2013(36):103-106.
[35] Zheng Jianming,Gu Meng,Xiao Jie,et al.Mitigating voltage fade in cathode materials by improving the atomic level uniformity of elemental distribution[J].Nano Lett.,2014,14(5):2628-2635.
[36] Hy S,Cheng Juhsiang,Liu Yue,et al.Understanding the role of Ni in stabilizing the lithium-rich high-capacity cathode material Li[NixLi(1-2x)/3Mn(2-x)/3]O2(0<=x<=0.5)[J].Chem.Mater.,2014(26):6919-6927.
猜你喜欢
晚晴(2020年7期)2020-12-03
理科考试研究·高中(2019年7期)2019-09-17
广东教育·高中(2018年12期)2018-02-13
分析化学(2017年12期)2017-12-25
世界家苑(2017年6期)2017-08-27
试题与研究·高考理综化学(2016年4期)2017-03-28
中国管理信息化(2016年21期)2016-12-27
中国高新技术企业(2015年27期)2015-07-30
电子技术与软件工程(2015年6期)2015-04-20
