
新型含吡唑基1,2,4-三唑衍生物的合成*
2015-04-18 07:11:27郑玉国郭晴晴王清河张国义兴义民族师范学院绿色化学合成技术研究所民族药用生物资源研究与开发实验室贵州兴义562400
合成化学 2015年8期
郑玉国, 郭晴晴, 王清河, 文 青, 周 莉, 张国义(兴义民族师范学院 绿色化学合成技术研究所 民族药用生物资源研究与开发实验室,贵州 兴义 562400)
·快递论文·
新型含吡唑基1,2,4-三唑衍生物的合成*
郑玉国, 郭晴晴, 王清河, 文 青, 周 莉, 张国义
(兴义民族师范学院 绿色化学合成技术研究所 民族药用生物资源研究与开发实验室,贵州 兴义 562400)
以1-苯基-3-甲基-5-吡唑啉酮为起始原料,经氯甲酰化、氧化、酯化、肼解、成盐及闭环反应制得4-氨基-5-(1-苯基-3-甲基-5-氯吡唑)-1,2,4-三唑-3-硫酮(6); 6与苯甲醛经缩合反应制得4-苯甲亚胺基-5-(5-氯-3-甲基-1-苯基)吡唑-4H-1,2,4-三唑-3-硫酮(7); 7与对硝基苄氯反应合成了2个新型含吡唑基1,2,4-三唑化合物,其结构经1H NMR, IR和元素分析表征。
吡唑啉酮; 1,2,4-三唑衍生物; 合成
1,2,4-三唑是易降解、毒性低、杀菌、杀虫、抗病毒和除草的农药活性基团,在农药领域具有良好的开发前景[1-8]。目前已开发出了三唑酮、氨唑草酮和三唑磷等数十个高效低毒的1,2,4-三唑农药品种。吡唑杂环化合物在植物体内具有良好的内吸传导性能,结构多样易于修饰。该类化合物显示出良好的生物活性,如具有止痛、抗炎、抗菌、抗肿瘤、调节植物生长、杀虫和抗病毒等药理活性[9-13]。目前已有呋吡菌胺、吡噻菌胺和吡虫酰胺等几十个吡唑药物已商品化。
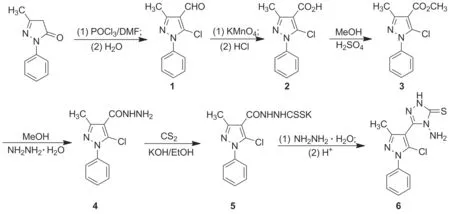

Scheme 1
本课题组[14-15]曾报道1,2,4-三唑类衍生物的合成,并对其生物活性进行了初步研究,取得了一定成果。在此基础上,本文以1-苯基-3-甲基-5-吡唑啉酮为起始原料,经氯甲酰化、氧化、酯化、肼解、成盐和闭环反应制得4-氨基-5-(1-苯基-3-甲基-5-氯吡唑)-1,2,4-三唑-3-硫酮(6); 6与苯甲醛经缩合反应制得4-苯甲亚胺基-5-(5-氯-3-甲基-1-苯基)吡唑-4H-1,2,4-三唑-3-硫酮(7); 7与对硝基苄氯反应合成了2个新型的含吡唑基1,2,4-三唑化合物——3-(4-硝基苯甲硫基)-N-苯次甲基-5-(5-氯-2-甲基-4-苯基吡唑)-4H-1,2,4-三唑-4-胺(8)和3-(5-氯-2-甲基-4-苯基吡唑)-6-苯基-7-(4-硝基苯基)-5H-1,2,4-三唑[3,4-b][1,3,4]噻二嗪(9)(Scheme 1),其结构经1H NMR, IR和元素分析表征。
1 实验部分
1.1 仪器与试剂
X-5型熔点仪(温度未校正);JEOL-ECX 400 Hz型核磁共振仪(DMSO-d6为溶剂,TMS为内标);IR Prestige-21型红外光谱仪(KBr压片);Elementar Vario-Ⅲ型元素分析仪。
所用试剂均为分析纯。
1.2 合成
(1) 1的合成
在反应瓶中加入DMF 27 mL,搅拌下于0 ℃缓慢滴加POCl3153 g(1.0 mol ),滴毕,缓慢加入1-苯基-3-甲基-5-吡唑啉酮20.9 g(0.12 mol),加毕,于80 ℃反应5 h[TLC监测,展开剂:A=V(石油醚) ∶V(乙酸乙酯)=1 ∶1]。冷却至室温,缓慢倒入100 mL冰水中,静置2 h;抽滤,滤饼干燥,用无水乙醇重结晶得淡黄色固体3-甲基-1-苯基-5-氯-4-甲醛基吡唑(1),收率82%, m.p.142 ℃~144 ℃。
(2) 2的合成
在反应瓶中加入1 11.1 g(0.05 mol)和水100 mL,搅拌下于室温反应5 min;缓慢滴入KMnO411.85 g(0.075 mol)水(200 mL)溶液,滴毕,于70 ℃反应8 h(TLC检测,展开剂:A=1 ∶1)。冷却,用10%KOH溶液调至pH 10,过滤,滤液用浓盐酸酸化,抽滤,滤饼干燥,用甲醇重结晶得白色粉末3-甲基-1-苯基-5-氯-4-吡唑甲酸(2),收率93%, m.p.240 ℃~242 ℃。
(3) 3的合成
在反应瓶中加入2 118.5 g(0.5 mol)和无水甲醇250 mL,搅拌下于室温反应5 min;缓慢滴入浓硫酸10 mL,滴毕,回流反应8 h(TLC检测,展开剂:A=1 ∶1)。冷却,旋蒸脱溶,用饱和NaHCO3溶液洗涤(有白色固体析出),抽滤,滤饼用无水乙醇重结晶得白色针状晶体1-苯基-3-甲基-5-氯-4-吡唑甲酸甲酯(3),收率65.7%, m.p.179 ℃~180 ℃。
(4) 4的合成
在反应瓶中依次加入3 12.5 g(0.05 mol),无水甲醇10 mL和80%水合肼20 mL,搅拌下回流反应5 h(TLC检测,展开剂:A=1 ∶1)。冷却,抽滤得白色片状固体1-苯基-3-甲基-5-氯-4-吡唑酰肼(4),收率74.2%, m.p.164 ℃~166 ℃。
(5) 5的合成
在反应瓶中依次加入4 6.28 g(0.025 mol),无水乙醇200 mL和氢氧化钾1.4 g(0.025 mol),搅拌下于室温反应5 min;缓慢滴加二硫化碳2.28 g(0.030 mol),滴毕,反应12 h。抽滤,滤饼用无水乙醇洗涤,干燥得白色固体N-(1-苯基-3-甲基-5-氯)吡唑甲酰肼基二硫代甲酸钾(5),收率75.2%, m.p.>250 ℃。
(6) 6的合成
在反应瓶中加入5 3.65 g(0.010 mol)和80%水合肼30 mL,搅拌下回流反应2 h(TLC检测,展开剂:A=3 ∶1)。自然冷却至室温,过滤,滤液用10%盐酸调至中性(析出固体),抽滤,滤饼用水洗涤,干燥后用无水乙醇重结晶得白色粉末6,收率65.8%, m.p.266 ℃~268 ℃;1H NMRδ: 14.01(s, 1H, NH), 7.62~7.53(m, 5H, PhH), 5.65(s, 2H, NH2), 2.29(s, 3H, CH3); IRν: 3 436, 3 253, 3 064, 1 632, 1 596, 1 453, 1 363 cm-1; MSm/z: 307.18{[M+H]+}; Anal.calcd for C12H11N6SCl: C 46.98, H 3.61, N 27.39; found C 47.12, H 3.83, N 27.66。
(7) 7的合成
在反应瓶中依次加入无水乙醇30 mL, 6 3.07 g(0.010 mol)和苯甲醛1.27 g(0.012 mol),搅拌使其溶解;加入冰醋酸5滴,于65 ℃反应5 h(TLC检测,展开剂:A=5 ∶1)。冷却,抽滤,滤饼用水洗涤,干燥后用无水乙醇重结晶得白色片状固体7,收率72%, m.p. 215 ℃~217 ℃;1H NMRδ: 14.39(s, 1H, NH), 9.99(s, 1H, NCH), 7.86~7.50(m, 10H, PhH), 2.30(s, 3H, CH3); IRν: 3 108, 2 950, 2 550, 1 614, 1 601, 1 590, 1 560, 1 470, 1 372, 1 279, 822 cm-1; MSm/z: 395.22{[M+H]+}; Anal.calcd for C19H15N6SCl: C 57.79, H 3.83, N 21.28; found C 57.83, H 3.97, N 21.42。
(8) 8的合成
在反应瓶中依次加入DMF 2 mL和7 0.40 g(0.001 mol),搅拌使其溶解;依次加入蒸馏水30 mL和3%氢氧化钠水溶液10 mL,加毕,于室温反应10 min;加入对硝基氯化苄0.21 g(1.2 mmol),反应24 h(TLC检测,展开剂:A=1 ∶1)。分液,水相用乙酸乙酯(4×30 mL)萃取,合并萃取液,用无水MgSO4干燥,浓缩后经硅胶柱层析(洗脱剂:A=3 ∶1)纯化得白色粉末8,收率60.3%, m.p.172 ℃~174 ℃;1H NMRδ: 8.79(s, 1H, N=CH), 8.14(d,J=8.8 Hz, 2H, PhH), 7.76(d,J=8.8 Hz, 2H, PhH), 7.65~7.50(m, 10H, PhH), 4.56(s, 2H, SCH2), 2.27(s, 3H, CH3); IRν: 3 010, 2 931, 1 616, 1 589, 1 456, 1 436, 660 cm-1; Anal.calcd for C26H20N7O2SCl: C 58.92, H 3.80, N 18.50; found C 59.13, H 3.94, N 18.67。
(9) 9的合成
在反应瓶中依次加入7 0.40 g(1 mmol), DMF 50 mL和K2CO30.1 g(1 mmol),搅拌下加入对硝基氯化苄0.21 g(1.2 mmol),于100 ℃反应3 h(TLC检测,展开剂:A=1 ∶1)。过滤,滤液减压蒸馏,残余物经硅胶柱层析(洗脱剂:A=3 ∶1)纯化得白色片状固体9,收率67.2%, m.p.206 ℃~208 ℃;1H NMRδ: 8.10(d,J=8.8 Hz, 2H, PhH), 7.74(d,J=8.8 Hz, 2H, PhH), 7.57~7.23(m, 11H, PhH, NH), 5.25(d,J=10.4 Hz, 1H, SCH), 4.99(t,J=10.4 Hz, 1H, NCH), 2.27(s, 3H, CH3); IRν: 3 159, 3 004, 2 950, 1 590, 1 524, 1 483, 669 cm-1; Anal.calcd for C26H20N7O2SCl: C 58.92, H 3.80, N 18.50; found C 59.21, H 3.99, N 18.77。
2 结果与讨论
2.1 合成
1的合成通过Vilsmeier-Haack氯甲酰化反应引入醛基,POCl3既作为溶剂又作反应试剂。
在2的合成中,需缓慢逐滴滴加KMnO4水溶液,以防止反应放热温度急剧升高。
在4的合成中,需逐滴滴入80%水合肼,防止吡唑环5-Cl被NH2NH2取代,引起副反应发生。
在5的合成中,CS2需缓慢滴入,防止反应剧烈将吡唑苯甲酰肼包夹从而导致反应不完全。
在7的合成中,加入冰乙酸作为催化剂,提供适宜的H+,生成羰基盐正离子,更容易发生亲核加成反应。
7在NaOH水溶液中转化为亲核性更强的硫负离子,DMF作助溶剂,经亲核取代反应合成8。
以K2CO3为缚酸剂,DMF为溶剂,7与对硝基氯化苄发生亲核取代加成反应合成9。
8中NO2的强吸电子作用使得CH2变活泼,推测其在加热条件和K2CO3催化下,可与C=N双键发生分子内加成闭环转化为9。
2.2 表征
1H NMR分析表明,6转变为7后,NH2吸收峰消失,在低场(δ9.99)出现亚胺质子吸收峰。 7转变为8后,NH吸收峰消失,在δ4.56出现SCH2吸收峰;7转变为9后, N=CH吸收峰消失,在δ5.25处出现SCH质子吸收峰,其受N-CH偶合裂分为二重峰, 耦合常数为10.4 Hz;在δ4.99处出现N-CH质子吸收峰,与NH及SCH偶合表现为三重峰, 耦合常数10.4 Hz。
IR分析表明,6转变为7后,NH2伸缩振动吸收峰消失,出现C=N(1 614 cm-1)吸收峰。7转化为8和9后,新出现C-S-C伸缩振动强吸收峰。8中由于吡唑及三唑环的共轭效应,通过链传递致使C=N的电子云密度降低,因而向高场移动,在1 616 cm-1出现C=N特征峰,在660 cm-1处新出现C-S-C伸缩振动强吸收峰。9中,在669 cm-1处新出现C-S-C伸缩振动强吸收峰,N-H伸缩振动吸收峰出现在3 159 cm-1。
[1] 魏学,郑玉国,薛伟,等. 新型1,2,4-三唑并[3,4-b]-1,3,4-噻二唑类衍生物合成及抗病毒活性[J].合成化学,2010,18(5):595-598.
[2] 陈文彬,金桂玉.α-[(1H-1,2,4-三唑-1-基)甲基]-β-芳基-β-芳硫基取代苯丙醇的合成及生物活性研究[J].应用化学,2002,19(6):527-530.
[3] 鲍小平,熊启中,邹林波,等. 2-苄硫基-5-甲基-1,2,4-三唑并[1,5-a]嘧啶-7-氧乙酰腙类衍生物的合成及其抑菌活性[J].合成化学,2013,21(1):53-57.
[4] Li F L, Dai B, Song H B,etal. Synthesis,structure,and fungicidal activity of triorganotin (4H-1,2,4-triazol-4-yl)benzoates[J].Heteroat Chem,2009,20(7):411-417.
[5] Wang B L, Shi Y X, Ma Y,etal. Synthesis and biological activity of some novel trifluoromethyl-substituted 1,2,4-triazole and bis(1,2,4-triazole) Mannich bases containing piperazine rings[J].J Agric Food Chem,2010,58(23):5515-5522.
[6] Wang M H, Zhu R Z, Fan Z J,etal. Bitriazolyl acyclonucleosides synthesized via Huisgen reaction using internal alkynes show antiviral activity against tobacco mosaic virus[J].Bioorg Med Chem Lett,2011,21(1):354-357.
[7] 杜婕,杜海棠,桑维钧,等. 新型3-(3,4,5-三甲氧基苯基)-4-氨基-5-取代苄砜基-1,2,4-三唑的合成及其抑菌活性[J].合成化学,2014,22(4):485-488.
[8] 于赛男,李智超,胡英芝,等. 含三氟甲基和双希夫碱化合物的合成与表征[J].合成化学,2010,18(2):226-228.
[9] 杨金凤,陶晶,李炳奇,等. 芳基吡唑腙及其双杂环化合物的合成与抗菌活性[J].合成化学,2009,17(2):151-154.
[10] 刘新华,白林山,王世范,等. 5-(2-羟基苯基)-3-甲基吡唑酰胺衍生物的合成与杀菌活性[J].合成化学,2006,14(2):147-149.
[11] 闫启东,徐俊,徐峰,等. 1-[6-(3,5-二甲基-1H-吡唑-1-基)-1,2,4,5-四嗪-3-基]酰肼及其衍生物的合成与表征[J].合成化学,2011,19(6):709-713.
[12] 廖国辉,张阳,魏宁宁,等. 1-甲基-3-二氟甲基-4-吡唑酰胺类衍生物的合成及其抑菌活性[J].合成化学,2011,19(1):19-23.
[13] 王京,张磊,姚其正. 新型含1,3,4-噁二唑的吡唑类化合物的合成及其抗肿瘤活性[J].合成化学,2014,22(6):730-733.
[14] 郑玉国,魏学,郭晴晴,等. 双三唑席夫碱哒嗪酮衍生物合成及其生物活性[J].应用化学,2011,28(9):1028-1034.
[15] 郑玉国,薛伟,郭晴晴,等. 三唑席夫碱苯并吡喃酮衍生物合成及其抗病毒活性[J].合成化学,2012,20(3):316-319.
Synthesis of Novel 1,2,4-Triazolo Derivatives Containing Pyrazole Unit
ZHENG Yu-guo, GUO Qing-qing, WANG Qing-he, WEN Qing, ZHOU Li, ZHANG GUO-yi
(Institute of Green Chemistry-Synthesis Technology, Key Laboratory of Research and Development of Ethnomedicinal Biological Resources, Xingyi Normal University for Nationalities, Xingyi 562400, China)
4-Amino-5-(5-chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-2H-1,2,4-triazole-3(4H)-thione(6) was afforded in six steps including chloromethylation, oxidation, esterification, hydrazidation, salt formation and cyclization from 3-methyl-1-phenyl-1H-pyrazol-5(4H)-one. 4-Benzylideneamino-5-(5-chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-2H-1,2,4-triazole-3(4H)-thione(7) was then synthesized by condensation reaction of 6 with benzaldehyde. Two novel 1,2,4-triazolo derivatives containing pyrazole unit were synthesized by reaction of 7 with 1-(chloromethyl)-4-nitrobenzene. The structures were characterized by1H NMR, IR and elemental analysis.
pyrazolone; 1,2,4-triazolo derivative; synthesis
2015-02-09
贵州省教育厅自然科学研究项目(黔教科2010090); 贵州省高等学校教学质量与教学改革工程资助项目(黔教高发[2012]426号和[2013]446号); 黔西南州科技计划项目(2013-6)
郑玉国(1982-),男,汉族,山东寿光人,副教授,主要从事新农药的创制研究。 Tel.0859-3568054, E-mail: yuguobaolong@163.com
O621.3; O626
A
10.15952/j.cnki.cjsc.1005-1511.2015.08.0729
猜你喜欢
今日农业(2021年2期)2021-11-27 19:19:53
今日农业(2020年23期)2020-12-31 09:00:40
中国化工贸易·中旬刊(2019年10期)2019-10-21 17:46:46
中国果菜(2016年9期)2016-03-01 01:28:41
合成化学(2015年1期)2016-01-17 08:59:30
西华师范大学学报(自然科学版)(2015年3期)2015-02-27 15:31:19
天然产物研究与开发(2014年3期)2014-04-27 14:15:32
火炸药学报(2014年5期)2014-03-20 13:17:47
中国蔬菜(2014年3期)2014-02-01 18:06:17
化学分析计量(2011年1期)2011-04-11 13:13:24
