
普乐沙福的合成工艺改进*
2015-04-23 10:55:38陈贝贝卓广澜
合成化学 2015年8期
陈贝贝,强 斌,卓广澜
(浙江理工大学化学系,浙江 杭州 310018)
普乐沙福(Plerixafor),化学名为 1,1'-[1,4-亚苯基二(亚甲基)]-二-1,4,8,11-四氮杂环十四烷,是美国Genzyme公司研发的趋化因子受体4(CXCR4)专一性拮抗剂,为一种提升造血干细胞数量的药物,商品名为 Mozobil。美国 FDA于2008年12月首次批准该药物上市,主要用于接受干细胞骨髓移植的多发性骨髓瘤和非霍奇金淋巴瘤等成年肿瘤患者[1-2]。
有关Plerixafor的合成方法主要有二种。方法一:以丙烯酸甲酯为起始原料,先与乙二胺Michael加成、胺解,再与丙二酸二甲酯缩合制得1,4,8,11-四氮杂-5,7,12-三氧代-环十四烷,最后经α,α-二溴对二甲苯桥连还原得 Plerixafor[3]。方法二:以1,4,8,11-四氮杂环十四烷(5)为原料,经保护(如磷酰基[4]、对甲苯磺酰基[5]和三氟乙酰基[6]等)、与 α,α-二溴对二甲苯桥连,最后脱保护制得Plerixafor。方法一采用无水乙二胺及无水四氢呋喃,工艺条件较苛刻,同时还用到硼烷二甲硫醚,反应选择性和收率均不高;方法二中5原先不易制备,价格高,但通过本研究方法,可大量制备得到,从而解决了该方法的应用瓶颈。
本文综合文献[5-9]方法,以二(3-氨基丙基)乙二胺(1)经保护、环合和脱保护反应制得关键中间体5;5与三氟乙酸乙酯反应制得三保护化合物6;6在碳酸钾作用下与α,α-二溴对二甲苯桥连制得化合物7;7在碳酸钾作用下脱保护合成了Plerixafor(Scheme 1),总收率30%,其结构经1H NMR和ESI-MS确证。并对合成工艺进行改进。
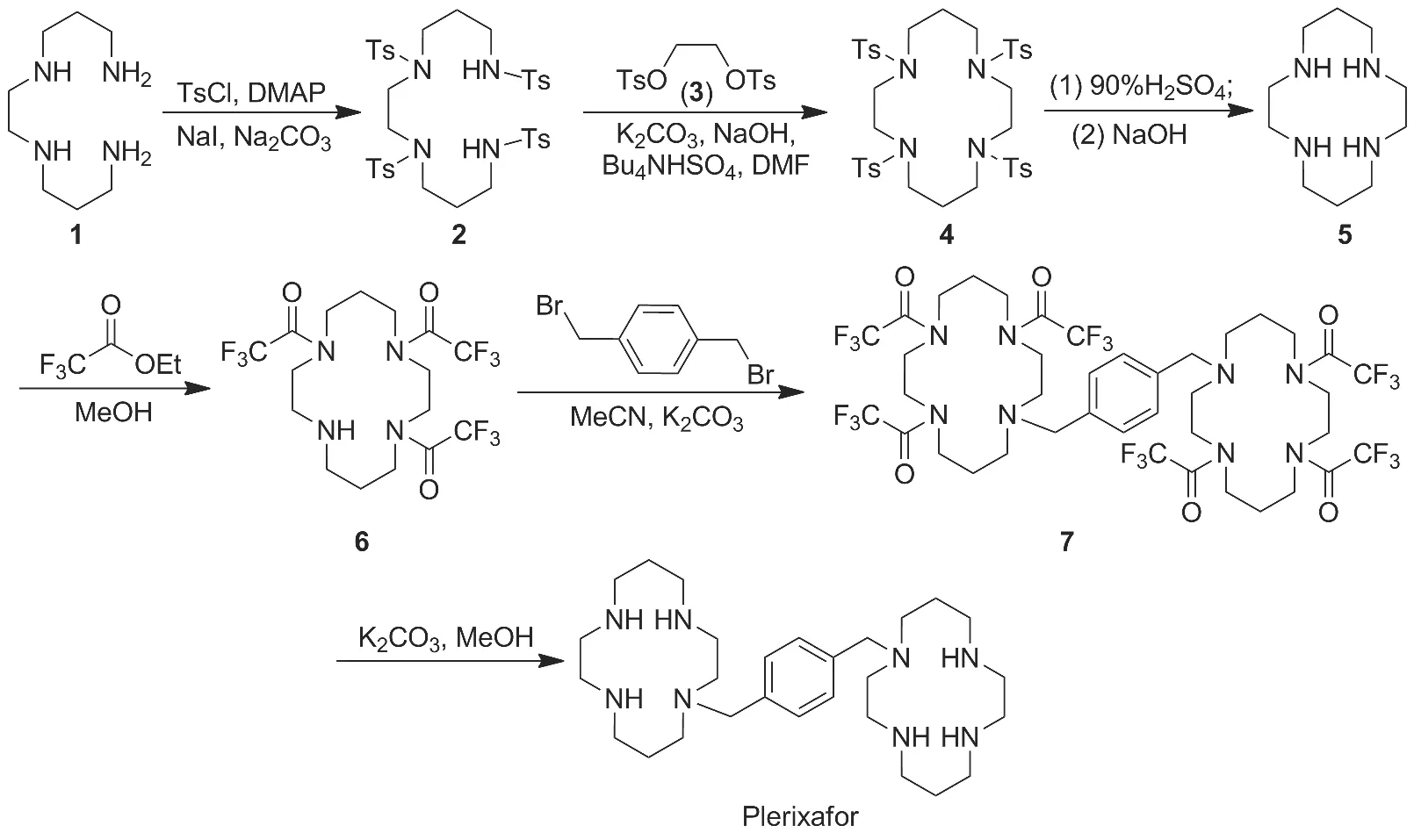
Scheme 1
1 实验部分
1.1 仪器与试剂
B-540型电热熔点仪(温度未校正);Varian Unity NOYA-400/54型核磁共振仪(CDCl3为溶剂,TMS为内标);天美GC7900型气相色谱仪;Agilent 6890N/5973I型气相色谱质谱联用仪。
碳酸钠,化学纯;其余所用试剂均为分析纯。
1.2 合成
(1)2的合成
在反应瓶中依次加入 1 5.00 g(28.68 mmol),碳酸钠12.72 g(120.00 mmol),4-二甲氨基吡啶(DMAP)17 mg(0.14 mmol),碘化钠 0.02 g(0.14 mmol)和水115 mL,搅拌下水浴升温至60℃,保持温度不超过65℃缓慢滴加对甲苯磺酰氯23.50 g(123.23 mmol)的THF(40 mL)溶液,滴毕(约1.5 h),反应2 h。冷却至55℃,静置分液,下层澄清液用混合溶剂[正庚烷10 mL+THF 10 mL]萃取,合并萃取液,升温至45℃,滴加0.57 mol·L-1盐酸6.3 mL,反应5 min。加入正庚烷 25 mL,冷却至室温,抽滤,滤饼依次用水和乙醇洗涤,真空干燥得白色固体2 18.6 g,收率82%,m.p.160 ℃ ~162 ℃(162 ℃ ~164 ℃[10]);1H NMR δ:1.8(m,4H),2.42(s,6H),2.44(s,6H),2.98(s,4H),3.16(t,J=6.57 Hz,4H),3.25(s,4H),5.28(s,2H),7.26 ~7.34(m,8H),7.66 ~7.74(m,8H);ESI-MS m/z:792{[M+H]+}。
(2)4的合成
在反应瓶中依次加入 2 10.0 g(12.64 mmol),氢氧化钠 1.80 g(45 mmol),碳酸钾 2.10 g(15.21 mmol)和 DMF 180 mL,油浴升温至 80℃,反应3 h。加入四丁基硫酸氢铵0.4 g(1.18 mmol),升温至100℃,滴加1,2-二(对甲苯磺酰氧基)乙烷(3)6.60 g(17.82 mmol)的 DMF(120 mL)溶液,滴毕(2 h),反应2 h。冷却至室温,搅拌过夜,冰浴冷却下加水(250 mL)淬灭反应,过滤,滤饼用水洗涤,抽干,加入乙腈120 mL,搅拌下回流2 h;冷至室温,抽滤,滤饼用乙腈洗涤,真空干燥得白色固体4 7.6 g,收率73%,m.p.284 ℃ ~286 ℃(288 ℃ ~289 ℃[10]);1H NMR δ:1.87(m,4H),2.44(s,12H),3.14(t,J=6.5 Hz,8H),3.22(s,8H),7.34(d,J=7.9 Hz,8H),7.70(d,J=7.8 Hz,8H);ESI-MS m/z:818{[M+H]+}。
(3)5的合成
在反应瓶中依次加入 4 11.00 g(13.46 mmol)和90%硫酸33 mL,搅拌下于100℃反应48 h。冷却至2℃,依次滴加无水乙醇120 mL和乙醚100 mL(控制温度不超过10℃),滴毕,抽滤,滤饼依次用少许乙醇、乙醚洗涤,真空干燥得灰白色固体,用水(60 mL)溶解,滴加2 mol·L-1氢氧化钠溶液(50 mL),滴毕,减压蒸除水,残余物加入氯仿250 mL,剧烈搅拌,抽滤,滤饼用少许氯仿润洗,滤液用无水硫酸钠干燥后浓缩得白色固体 5 2.44 g,收率 90%,m.p.182 ℃ ~183 ℃(185 ℃ ~ 186 ℃[10]);1H NMR δ:1.75(m,4H),2.15(s,4H),2.70(s,8H),2.77(t,J=5.3 Hz,8H);ESI-MS m/z:201{[M+H]+}。
(4)6的合成
在反应瓶中依次加入5 7.5 g(37.5 mmol),三乙胺5.4 mL和无水甲醇35 mL,搅拌使其溶解;冰水浴冷却,快速滴加三氟乙酸乙酯21.25 g(134.4 mmol),滴毕(约5 min),于室温反应 6 h。减压浓缩,残余物经快速硅胶柱层析(洗脱剂:乙酸乙酯)纯化得泡沫状白色固体6 16.1 g,收率88%;1H NMR δ:1.20 ~1.40(m,2H),1.65 ~1.95(m,2H),2.60 ~ 2.80(m,2H),2.88 ~3.05(m,2H),3.40 ~ 3.85(m,12H);ESI-MS m/z:489{[M+H]+}。
(5)7的合成
在反应瓶中依次加入6 3.24 g(6.64 mmol),无水碳酸钾 2.75 g(19.92 mmol),α,α-二溴对二甲苯0.88 g(3.32 mmol)和无水乙腈20 mL,搅拌下回流反应5 h。冷却至室温,抽滤,滤液减压蒸除溶剂,用80%乙醇(20 mL)重结晶,真空干燥得白色 固体72.36 g,收 率 66%;1H NMR δ:1.63~1.85(m,4H),2.01 ~ 2.56(m,8H),2.75(s,4H),3.31 ~ 3.78(m,28H),7.16(s,4H);ESI-MS m/z:1 079{[M+H]+}。
(6)Plerixafor的合成
在反应瓶中依次加入7 2.20 g(2.04 mmol),无水碳酸钾1.13 g(8.16 mmol)和无水甲醇10 mL,搅拌下回流反应24 h。冷却至室温,加入甲苯20 mL,浓缩至约15 mL,加入甲苯30 mL,升温至50℃,趁热抽滤,滤饼用少许甲苯润洗,合并滤液和洗液,减压蒸除溶剂得白色固体Plerixafor 0.98 g,收率 96%,m.p.125 ℃ ~128 ℃;1H NMR δ:1.62 ~ 1.81(m,8H),2.46 ~ 2.75(m,32H),3.55(s,4H),7.37(s,4H);ESI-MS m/z:503{[M+H]+}。
2 结果与讨论
2.1 合成
为了提高收率和适于工业放大,本文进行了相关工艺改进。
在以1和对甲苯磺酰氯为原料合成2的过程中,尝试了三种方法:方法一以氢氧化钠为碱,乙醚和水为溶剂;方法二以碳酸钾为碱,水为溶剂;方法三以碳酸钠为碱,同时加入催化量碘化钠和DMAP,四氢呋喃和水为溶剂。实验结果表明:方法一收率78.0%,方法二收率75.2%,方法三收率81.9%。说明采用适量的催化剂及选用碳酸钠作碱,在四氢呋喃和水混合溶剂中的反应效果最好,且反应时间较短,后处理无需柱层析纯化,用无水乙醇加热搅拌即可提纯。推测催化量的碘化钠可以活化对甲苯磺酰氯上的Cl原子,使得其加速其离去,DMAP作为缚酸剂,有助于引发反应。本文选择方法三合成2。

表1 反应条件对2收率的影响*Table 1 Effect of reaction conditions on yield of 2
在4的合成中,2 12.64 mmol,其余反应条件同1.2(2),考察原料配比[r=n(2)∶n(3)]对4收率的影响,结果见表2。由表2可见,在r由1 ∶1变为 1 ∶1.4 的过程中,随 1,2-二(对甲苯磺酰氧基)乙烷比例增加,环化反应会趋于反应完全,收率由 59.3% 提高至 73.8%;当 1,2-二(对甲苯磺酰氧基)乙烷用量继续增加,收率基本不变。因此最佳 r为1∶1.4。

表2 r对4收率的影响*Table 2 Effect of r on yield of 4
在6的合成中,尝试多种保护剂。实验结果表明,用对甲苯磺酰基保护时收率不高;磷酰基保护时可克服过度取代,但反应要求绝对无水条件,采用一锅法制备则面临无法控制投料比及进程不易跟踪等问题;改用三氟乙酰基保护,通过控制投料比,可主要获得三保护产物,柱层析纯化只需用乙酸乙酯洗脱即可,操作简易,收率88%。
以三氟乙酸乙酯为保护剂,5 37.5 mmol,其余反应条件同1.2(4),进一步考察三氟乙酸乙酯用量对6收率的影响,结果见表3。由表3可见,当γ[n(5)∶n(三氟乙酸乙酯)]为1 ∶3时,TLC 检测显示有二保护化合物生成,收率78.6%,反应选择性不高;当γ=1∶4时,TLC检测显示无副产物生成,反应选择性好,收率较高(87.9%);当继续增加三氟乙酸乙酯用量至γ=1∶6时,收率略有下降(86.5%)。因此γ=1∶4较佳。

表3 γ对6收率的影响*Table 3 Effect of γ on yield of 6
7经脱保护合成Plerixafor的过程中,在碳酸钾作用下三氟乙酰基的脱除较易进行,反应条件温和,且不会产生大量废酸,减少环境污染。
综上所述,以二(3-氨基丙基)乙二胺为起始原料,经磺酰化、闭环、脱保护、碱化和桥连等反应合成了抗肿瘤药普乐沙福,总收率30%。该方法收率较高,适合工业化生产。
[1]De Clercq E.The bicyclam AMD3100 story[J].Nat Rev Drug Discov,2003,2(7):581 - 587.
[2]高源.Plerixafor(Mozobil)[J].中国药物化学杂志,2009,19(4):315.
[3]Achmatowicz M,Hegedus L S.Direct synthesis of 1,1'-[1,4-phenylenebis(methylene)]-bis-1,4,8,11-tetraazacyclotetradecane octahydrochloride(AMD 3100)without the use of protecting groups[J].J O rg Chem,2003,68(16):6435 -6436.
[4]Guillaume D,Marshall G R.Efficient one-pot synthesis of JM3100[J].Synth Commun,1998,28(15):2903-2906.
[5]苏靖,刘 瑶,郑志兵.Plerixafor的合成[J].中国医药工业杂志,2007,38(6):398 -400.
[6]Giandomenico C M,Yang W.Process for preparation of N-1 protected N ring nitrogen containing cyclic polyamines and products thereof[P].WO 2 002 026 721,2002.
[7]Ciszewski L,Amedio J,Kapa P,et al.Process for preparing 1,4,8,11-tetraazacyclotetradecane[P].WO 1 997 005 123,1997.
[8]Jacobson O,Weiss I D,Szajek L,et al.64Cu-AMD-3100——A novel imaging agent for targeting chemokine receptor CXCR4[J].Bioorganic & Medicinal Chemistry,2009,17(4):1486 -1493.
[9]Giandomenico C M,Sartori M,Moore D A.Facile N-1 protection of cyclam,cyclen and 1,4,7-triazacyclononane[J].Tetrahedron Letters,2003,44(12):2481-2483.
[10]Rubio M,Astorga C,Alfonso I,et al.Chemoenzymatic syntheses of polyamines and teraazamacrocycles[J].Synth Commun,2002,32(16):2441-2452.
猜你喜欢
河南畜牧兽医(2021年9期)2021-12-10 10:43:50
——非均布滤饼的局部比阻与平均比阻的测定与计算方法
化工装备技术(2020年4期)2020-09-09 07:34:10
流体机械(2020年5期)2020-06-24 05:39:08
杭州化工(2020年1期)2020-05-09 14:00:54
山东冶金(2019年6期)2020-01-06 07:46:02
石油钻探技术(2018年5期)2018-10-13 07:25:18
浙江化工(2017年4期)2017-05-11 02:37:40
中国矿业(2017年2期)2017-02-28 02:12:56
含能材料(2015年1期)2015-05-10 00:50:52
中国农资(2015年14期)2015-01-31 16:34:49
