
Evofosfamide的合成工艺改进*
2015-04-23 10:55:38陈冬寅
合成化学 2015年8期
姚 坤,陈冬寅,李 飞
(南京医科大学药学院,江苏 南京 211166)
氮芥类生物烷化剂是临床广泛使用的抗肿瘤药物。环膦酰胺和异环膦酰胺等氮芥膦酰胺类前药虽然能够降低氮芥的毒副作用,但是其安全性仍然不能令人满意[1]。重度乏氧是很多恶性肿瘤的特点,增加肿瘤细胞对放化疗的耐受性、促使肿瘤血管生成和肿瘤细胞转移,是重要的不良预后因子[2-3]。
Evofosfamide[(1-甲基-2-硝基-1H-咪唑-5-基)甲基-N,N-二(2-溴乙基)磷酰(1)]是2012年由默克公司研发的用于治疗晚期胰腺癌的药物,它是氮芥膦酰胺类潜药。在正常组织中,由于氧的浓度较高,该药物的硝基不被还原,从而以毒性较小的潜药状态存在。在肿瘤组织中,由于氧浓度较低,吸电子的硝基被还原为供电子的氨基,使药物不能稳定存在而被代谢,释放出强细胞毒性的氮芥类代谢产物,杀死肿瘤细胞。研究发现,1在缺氧条件下的细胞毒性远高于在有氧的条件下的细胞毒性,最高相差1 000倍左右[4-5]。1在乏氧组织中具有高选择性活性,它对远离血管的乏氧组织有非常好的杀伤力,而传统的抗癌药物如吉西他滨可以比较好地清除靠近血管的癌组织,因此,两者联合用药抗癌效果更佳[6]。将1与培美曲塞联合用药用于治疗晚期非鳞状非小细胞肺癌,疗效更佳[7]。人体中的细胞周期检查点激酶1抑制剂 AZD7762S能够增强1的药效[8]。目前,1已获得美国FDA批准进行多项临床试验,其中治疗胰腺癌和晚期软组织肉瘤已经进入Ⅲ期临床试验,并获得FDA批准进入快速通道[9]。
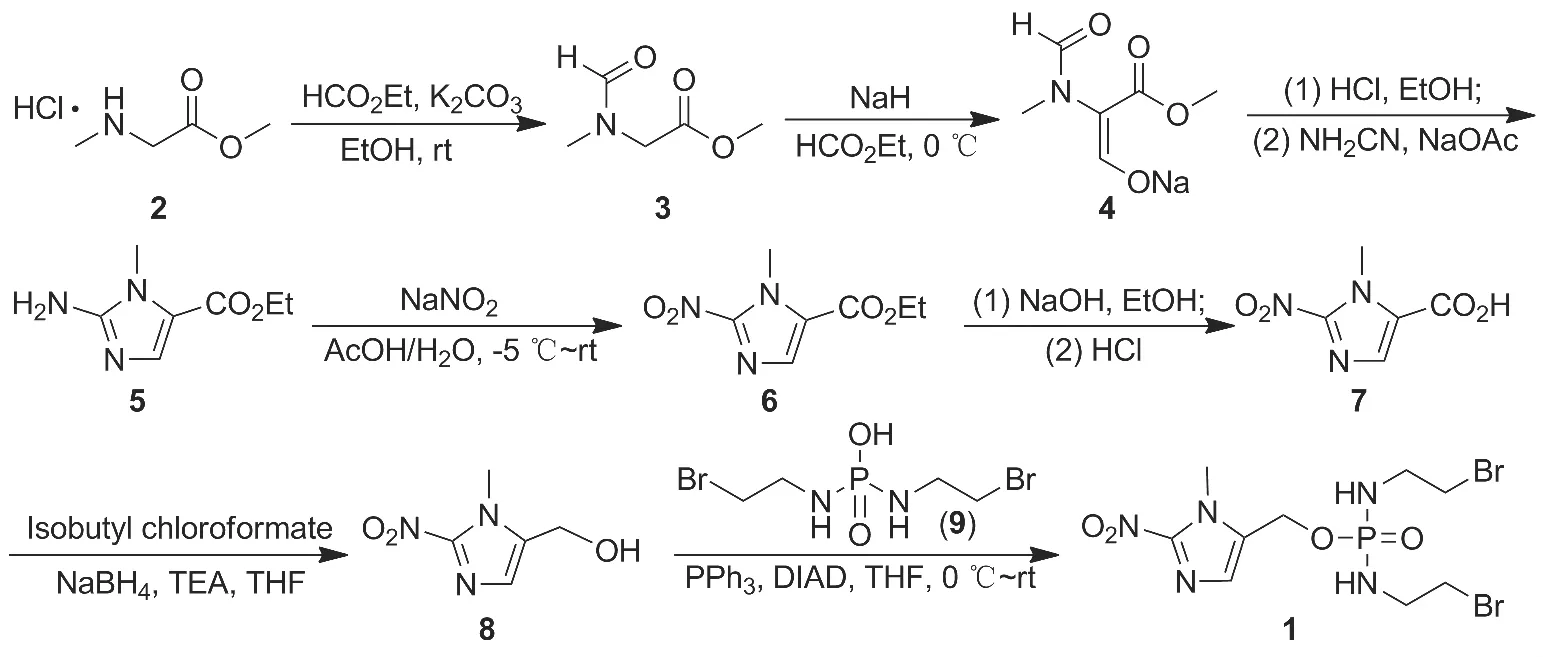
Scheme 1
2008年,Duan等[10]首次报道了1的合成路线。但文献中只简单介绍了关键中间体(3-甲基-2-硝基-3H-咪唑-4-基)甲醇(8)与氮芥类化合物(9)拼合制备1的方法。但8较为昂贵,迄今为止,其合成方法鲜有文献[11]报道。该方法以N-甲酰肌氨酸甲酯(3)为原料,经缩合、环合、氧化、水解和还原反应等5步反应制得8;8再与9拼合得1,总收率8%,且其中两步反应需柱层析分离,不适合工业化生产。因此,对1的合成工艺进行探索具有重要的意义。
为了建立1的有效合成方法,本文在文献[11]方法的基础上,以肌氨酸甲酯盐酸盐(2)为原料,与甲酸乙酯经过甲酰化和缩合两步反应制得钠(E)-3-甲氧基-2-(N-甲酰基)-3-氧代丙基-1-氨乙基-1-盐(4);4在氰胺和乙酸钠作用下环合得1-N-甲基-2-氨基咪唑-5-羧酸乙酯(5);5经氧化得1-N-甲基-2-硝基咪唑-5-羧酸乙酯(6);6在碱性条件下水解、再还原羧基制得关键中间体8;8与9经过Mitsunobu反应合成了1(Scheme 1),总收率16%,其结构经1H NMR,13C NMR和HR-ESIMS确证。并对3,5和6的合成工艺进行改进。
1 实验部分
1.1 仪器与试剂
Bruker ARX-300型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Agilent-1100型质谱仪。
薄层层析用GF254型硅胶,青岛海洋化工厂生产;其余所用试剂均为分析纯或化学纯。
1.2 合成
(1)3的合成
在反应瓶中加入甲酸乙酯90 mL(1.11 mol)和乙醇80 mL,搅拌下依次加入2 20 g(0.14 mol)和碳酸钾20 g,于35℃反应2 h。抽滤,滤液减压浓缩,干燥得淡黄色固体 3 15 g,产率 82%;1H NMR δ:8.62(s,1H),4.68(s,2H),3.68(s,3H),3.47(s,3H);HR-ESI-MS m/z:Calcd for C5H9NO3{[M+H]+}131.057 7,found 131.058 2。
(2)5的合成
在反应瓶中加入3 5 g(38.2 mmol)和甲酸乙酯16 mL(0.20 mol),搅拌使其溶解;冷却至0℃,分批加入 NaH 1.6 g(60%油状混悬液,40 mmol),加毕,于室温反应过夜。有黏稠物析出,用刮刀取出,置研钵中,加入己烷20 mL,研磨;不溶物用混合溶剂[EtOH(10 mL)+浓盐酸(6 mL)]溶解,于110℃搅拌反应1 h。冷却至室温,过滤,滤饼用乙醇洗涤。合并滤液和洗液,减压浓缩得深棕色油状物。加入混合溶液[NH2CN 3.5 g(83 mmol)+乙酸钠9 g+10%HOAc 20 mL]中,于80℃反应1 h。反应混合物减压浓缩至1/3,用20%碳酸钠溶液调至pH≈9。用乙酸乙酯萃取,合并有机相,用无水Na2SO4干燥。减压浓缩得棕灰色固体,用丙酮重结晶得灰白色固体粉末5 4.8 g,产率 75%;1H NMR δ:7.62(s,1H),4.20 ~4.10(m,2H),3.52(s,3H),1.53 ~1.50(m,3H);HR-ESI-MS m/z:Calcd for C7H11N3O2{[M -H]-}169.084 6,found 169.085 1。
(3)6的合成
在反应瓶中加入亚硝酸钠10 g(0.14 mol)和水30 mL,搅拌使其溶解;冰水浴冷却,滴加5 3.7 g(21.9 mmol)的乙酸(20 mL)溶液,滴毕,于室温反应过夜。用二氯甲烷萃取,合并有机相,用无水Na2SO4干燥。减压浓缩得微红色固体,用丙酮重结晶得淡棕色固体6 2.8 g,产率65%;1H NMR δ:8.03(s,1H),4.23 ~4.10(m,2H),3.73(s,3H),1.30 ~ 1.27(m,3H);HR-ESI-MS m/z:Calcd for C7H9N3O4{[M+H]+}199.058 8,found 199.059 3。
(4)7的合成
在反应瓶中加入 6 3.9 g(19.6 mmol),1 mol·L-1NaOH溶液60 mL和水20 mL,搅拌使其溶解;于室温搅拌至溶液变为透明浅棕色。用浓盐酸调至pH 1,用乙酸乙酯萃取,合并有机相,用无水Na2SO4干燥。减压浓缩得淡棕色固体7 3.2 g,产率 95%;1H NMR δ:11.50(br s,1H),8.17(s,1H),3.72(s,3H);HR-ESI-MS m/z:Calcd for C5H5N3O4{[M -H]-}171.027 5,found 171.028 0。
(5)8的合成
在反应瓶中加入7 3.1 g(18.0mmol)和THF 36 mL,搅拌使其溶解;加入三乙胺14 mL,冰盐浴冷却,滴加氯甲酸异丁酯3.8 mL(28.8 mmol),滴毕,反应1 h。于0℃加入硼氢化钠3.6 g,缓慢滴加水60 mL,滴毕(约需1 h),有固体析出。过滤,滤饼用THF洗涤,合并滤液和洗液,减压浓缩,残余物用乙酸乙酯重结晶得橙色固体8 2.5 g,产率88%;1H NMR δ:7.44(s,1H),4.63(s,2H),3.72(s,3H);HR-ESI-MS m/z:Calcd for C5H7N3O3{[M -H]-}157.048 2,found 157.048 7。
(6)1的合成
在反应瓶中加入8 3.6 g(22.9 mol)和 THF 100 mL,搅拌使其溶解;依次加入三苯膦3 g,9 1.8 g(5.7 mmol)和二异丙基偶氮二羧酸酯(DIAD)2 mL,于室温反应2 h。减压浓缩,残余物经快速硅胶柱层析[梯度洗脱剂:V(甲苯)∶V(丙酮)=3 ∶0 ~3 ∶7]纯化得米白色固体1 4.9 g,产率 48%;1H NMR δ:7.23(s,1H),5.04 ~ 5.12(m,2H),4.95(d,J=7.60 Hz,2H),3.94(s,3H),3.42(t,J=7.24 Hz,4H),3.01 ~ 3.17(m,4H);13C NMR δ:146.04,134.15(d,J=32.00 Hz),126.20,55.60,42.74,34.36,34.11(d,J=17.40 Hz);HR-ESI-MS m/z:Calcd for C9H16N5O4PBr2{[M - H]-}446.930 2,found 446.930 7。
2 结果与讨论
1的合成,文献[11]方法以3为起始原料,经6步反应合成。该方法存在反应操作繁琐,总产率较低(6%),合成过程中两步反应需硅胶柱色谱进行分离纯化等缺陷,不适合工业化生产。本文采用价格较为便宜的2为起始原料的方法,经7步反应合成1。
在3的合成中,通过考察反应温度对收率的影响,得出35℃为最佳反应温度,使收率由76%(25℃)[11]提高至82%,反应时间也缩短至2 h。
在5的合成中,将反应温度由95℃[11]降至80℃ ~85℃,减少了由于高温所带来的副产物,进而提高了收率。
在6的合成中,首先采用了文献[11]方法中的硅胶柱色谱分离法,但由于反应本身不完全,且6与5极性差别较小,硅胶柱色谱分离造成产物损失,降低收率。为此,本文分别选用了乙醇、乙酸乙酯、甲苯和丙酮等溶剂重结晶的方法代替硅胶柱色谱分离。实验结果表明:经丙酮重结晶的6,纯度得到提升,且降低了实验的繁琐程度。
经过对3,5和6的工艺改进,不仅总收率提高至16%,而且操作简单,更适合于工业化生产。
[1]Kumar D,Tewari-Singh N,Agarwal C,et al.Nitrogen mustard exposure of murine skin induces DNA damage,oxidative stress and activation of MAPK/Akt-AP1pathway leading to induction of inflammatory and proteolytic mediators[J].Toxicology Letters,2015,235(3):161-171.
[2]刘小艳,许新华.乏氧微环境与肿瘤治疗抗拒[J].广东医学,2013,34(23):3667 -3669.
[3]Gilkes D M,Semenza G L,Wirtz D.Hypoxia and the extracellular matrix:Drivers of tumour metastasis[J].Nat Rev Cancer,2014,14(6):430 -439.
[4]Yeh J J,Kim W Y.Targeting tumor hypoxia with hypoxia-activated prodrugs[J].Journal of Clinical Oncology,2015,33(13):1505 -1508.
[5]Liu Q,Sun J D,Wang J,et al.TH-302,a hypoxiaactivated prodrug with broad in vivo preclinical combination therapy efficacy:Optimization of dosing regimens and schedules[J].Cancer Chemotherapy and Pharmacology,2012,69(6):1487 -1498.
[6]Borad M J,Reddy S G,Bahary N,et al.Ranomized phase II trial of gemcitabine plus TH-302 versus gemcitabine in patients with advanced pancreatic cancer[J].Journal of Clinical Oncology,2015,33(13):1475 -1482.
[7]Goldman J,Belani C,Novello S,et al.Randomized,double-blind,placebo-controlled trial of evofosfamide(TH-302)in combination with pemetrexed in advanced non-squamous non-small cell lung cancer[J].Annals of Oncology,2015,26(Suppl 1):29 -44.
[8]Sun J D,Meng F Y,Liu Q,et al.Association between Chk1 inhibitor AZD7762-mediated modulation of pharmacodynamic biomarkers and potentiation of hypoxia-activated prodrug evofosfamide(TH-302)antitumor efficacy in a human tumor xenograft model[M].San Francisco,American Association for Cancer Research,2015.
[9]http://www.fiercebiotech.com/tags/threshold-pharmaceuticals.
[10]Duan J,Jiao H,Kaierman J,et al.Potent and highly selective hypoxia-activated achiral phosphoramidate mustards as anticancer drugs[J].Journal of Medicinal Chemistry,2008,51(8):2412 -2420.
[11]Matteucci M,Duan J,Jiao H,et al.氨基磷酸酯烷化剂前体药物[P].CN 200 680 030 082.8,2009.
猜你喜欢
酿酒科技(2022年8期)2022-08-20 10:25:04
化工管理(2021年7期)2021-05-13 00:46:24
中国农资(2016年1期)2016-12-01 05:21:23
食品界(2016年4期)2016-02-27 07:37:06
中国卫生标准管理(2015年14期)2016-01-15 02:58:31
烟草科技(2015年8期)2015-12-20 08:27:06
大连工业大学学报(2015年4期)2015-12-11 04:06:50
山东医药(2015年38期)2015-12-07 09:12:28
化工进展(2015年3期)2015-11-11 09:08:25
应用化工(2014年4期)2014-08-16 13:23:09
