
反相高效液相色谱法快速测定竹节参中齐墩果酸的含量
2015-04-14 02:03:26王黎王文娟高苏亚
应用化工 2015年5期
王黎,王文娟,高苏亚
(西安医学院 药学院,陕西 西安 710021)
竹节参别名竹节三七、竹节人参、竹根七等,为五加科人参属植物竹节参Panax japonicus C. A.Mey.的干燥根茎,始载于《本草纲目拾遗》,是人参属当中的一野生品种,民间将蒸熟的肉质根作滋补药,生晒根茎作止血药具有滋补强壮、散瘀止痛、止血祛痰等功效,临床上常用于病后虚弱、劳嗽咳血、跌打损伤等[1-2]。其化学成分以皂苷类为主,齐敦果酸为主要苷元[3]。目前,中国药典收载该药还未有含量测定项,仅对有效成分进行了薄层色谱鉴别。本文采用反相高效液相色谱法对竹节参中皂苷类成分齐敦果酸的含量进行快速测定,为竹节参药材的质量控制与评价提供了一定依据。
1 实验部分
1.1 原料与仪器
齐敦果酸对照品(98%);竹节参药材(产地云南),经西安医学院生药教研室冯永辉副教授鉴定为正品竹节参Panax japonicus C.A.Mey;甲醇,色谱纯;水为三重蒸馏水。
P-680 型高效液相色谱仪;UV-170 型紫外检测器;BSA224S-CW 型电子天平;SB-5200DT 型超声波清洗机。
1.2 溶液的制备
1.2.1 对照品溶液的制备 精密称定齐敦果酸对照品2.36 mg 于2 mL 量瓶中,加甲醇稀释至刻度,制成1.18 mg/mL 对照品溶液,0.45 μm 微孔滤膜过滤,制备对照品溶液。
1.2.2 供试品溶液的制备[4]精密称取干燥后的竹节参药材粉末1.0 g,以研钵研为粉末后,过60 目筛,置于具塞锥形瓶中,向其中加入50 mL 甲醇,称定重量,于功率250 W,频率50 kHz 的超声中放置40 min,称定后用甲醇补足所减失重量,摇匀,滤过;精密量取续滤液10 mL,回收溶剂至干,加25%盐酸20 mL、三氯甲烷25 mL 至残渣中,加热水解1.0 h,再用三氯甲烷振摇提取2 次,每次25 mL,合并提取液,回收溶剂至干,残渣加甲醇溶解并转移至10 mL量瓶中,加甲醇至刻度,摇匀,取续滤液,0.45 μm 微孔滤膜过滤,即得供试品溶液。
2 结果与讨论
2.1 色谱条件与系统适用性
色谱柱为Kromasil C18柱(150 mm ×4. 6 mm,5 μm),流动相为乙腈-水(93 ∶7),检测波长为215 nm,流速为1.0 mL/min,理论塔板数以齐墩果酸色谱峰计不低于5 000,见图1。

图1 反相高效液相色谱图Fig.1 RP-HPLC chromatogram
2.2 线性实验
精密吸取齐墩果酸对照品溶液2,4,6,8,10,15,20 μL,按上述色谱条件进样,测定其峰面积值,以峰面积积分值的常用对数(lgY)为纵坐标,以标准品进样量的常用对数(lgX)为横坐标进行回归分析(Y=7.415 7X-0.811 5,r=0.999 8),结果表明,齐墩果酸在2.36 ~23.60 μg/mL 范围内浓度与峰面积呈良好的线性关系。
2.3 精密度实验
取齐敦果酸对照品溶液,按上述色谱条件以进样量20 μL 重复进样6 次,取得齐墩果酸峰面积测定结果平均值为117.612,RSD 为1.00%。
2.4 稳定性实验
取竹节参供试品溶液,分别放置于0,2,4,6,8 h按上述色谱条件进样10 μL 进行测定,齐敦果酸峰面积的RSD 为0.91%,表明该样品溶液在8 h 内测定稳定。
2.5 重复性实验
精密称取同一批待测竹节参药材粉末5 份,分别按1.2.2 节方法制备为供试品溶液,按上述色谱条件进样10 μL 进行测定,所得齐敦果酸峰面积的RSD 为0.63%,表明本方法重复性良好。
2.6 回收率实验
精密称定竹节参药材粉末0.2 g(已知齐墩果酸含量48.28 mg/g)6 份,分别按已知含量的80%,100%,120%加入齐墩果酸对照品溶液,按1.2.2 节方法制备样品溶液并测定,计算得齐墩果酸平均回收率为98.86%,RSD 为0.41%,结果见表1。

表1 竹节参中齐墩果酸回收率实验(n=6)Table 1 Recoveries of Oleanolic acid in Panax japonicus
2.7 样品含量测定
取竹节参药材待测样品按1.2.2 节方法制备供试品溶液,按上述色谱条件进样20 μL 进行测定,按外标法计算含量,结果表明该产地云南的竹节参药材中齐墩果酸平均含量为48. 277 mg/g,结果见表2。
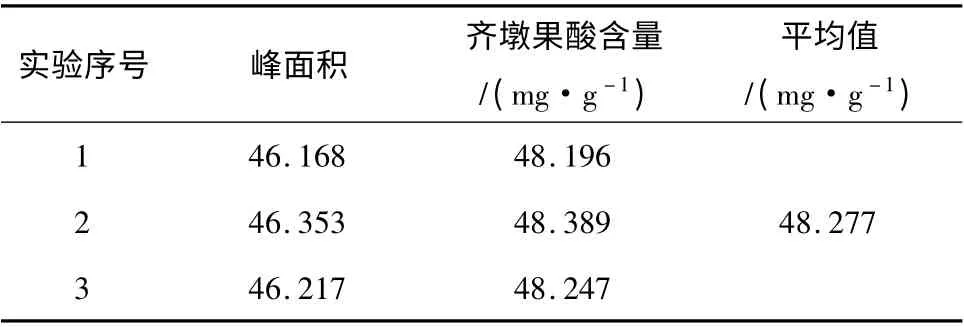
表2 待测云南产竹节参中齐墩果酸含量测定结果Table 2 The assay results of Oleanolic acid in Panax japonicus
3 讨论
3.1 检测波长的优化
采用UV 检测器对齐墩果酸对照品溶液进行全波长扫描,在210 nm 处出现最大吸收,但210 nm 检测时基线不平稳、溶剂峰干扰大、检测器响应值小。为了优化检测效果、减少干扰、提高信噪比,选择在215 nm 处检测,基线平稳且峰形满意。
3.2 流动相的优化
分别以甲醇-0.2%乙酸水溶液、甲醇-1%乙酸水溶液、甲醇-0.2%磷酸水溶液、甲醇-水为流动相来进行测定,结果此四种流动相体系所得的峰对称度较差,然后将其中的甲醇改为乙腈,当选用乙腈-水的流动相体系时,峰形较好。以乙腈-水的体积比分别为86∶14、88∶12、90∶10、93∶7 作流动相进行测定,发现流动相比例为86∶14 时峰形对称度较差,88∶12 时对照品后有一小峰且无法分开,90∶10 时峰形对称度有所改善但保留时间不够稳定(在6.0 ~7.5 min 之间波动),93∶7 时不仅峰形对称度高且分离效果较好、基线平稳,故将最终的流动相确定为乙腈-水(93∶7)。
4 结论
本文建立了以高效液相色谱法测定竹节参中三萜苷元齐墩果酸含量的方法,系统适用性实验表明选择乙腈-水的流动相体系符合中药材主要成分含量测定的要求;方法学考察表明该法快速准确、简单易行、重现性好,为竹节参药材的质量评价提供了一定依据。
[1] 王慧清. 中药材产销[M]. 成都:四川科学技术出版社,2004:320-322.
[2] 国家药典委员会.中国药典2005 年版[M].北京:化学工业出版社,2005:93.
[3] 国家中医药管理局《中华本草》编委会. 中华本草[M].第五卷.上海:上海科学技术出版社,1999:832-834.
[4] 袁丁,何毓敏,鲁科明,等. 中药竹节参中总皂苷和齐墩果酸的含量测定[J]. 西北药学杂志,2008,23(5):279-280.
猜你喜欢
食品与机械(2022年3期)2022-04-07 07:43:36
福建茶叶(2019年10期)2019-12-12 00:55:30
中成药(2018年10期)2018-10-26 03:40:56
天然产物研究与开发(2018年5期)2018-06-13 03:23:32
天然产物研究与开发(2016年1期)2016-06-05 10:29:24
医学研究杂志(2015年5期)2015-06-10 06:43:26
中国当代医药(2015年8期)2015-03-01 02:01:33
湖北科技学院学报(医学版)(2014年3期)2014-02-28 19:42:45
世界中医药(2014年10期)2014-02-07 02:16:28
