
DMAP 催化的17β-氰基-17α-羟基雄甾-4-烯-3-酮硅醚化反应研究
2015-04-14 02:03:12吴庆安吴红卫柏挺
应用化工 2015年5期
吴庆安,吴红卫,柏挺
(1.浙江工业大学 化学工程学院,浙江 杭州 310014;2.上海新华联制药有限公司,上海 201400)
17β-氰基-17α-羟基雄甾-4-烯-3-酮(I)是制备醋酸化合物S 和17-羟孕酮的一个重要中间体[1],而醋酸化合物S 和17-羟孕酮又可以用来进一步合成可的松、氢化可的松等皮质激素类药物。在化合物(I)的氰基上引入一个碳链,一般需要在强碱条件下进行,所以有必要将其17α-OH 加以保护。保护甾体羟基的方法很多,如酯化、醚化及硅醚化等[2-4]。通过比较,选择氯甲基二甲基氯硅烷(CDCS)作为硅醚化试剂对化合物(I)的17α-OH 进行保护。同时还可以达到另一个效果,即生成的17β-氰基-17α-羟基雄甾-4-烯-3-酮-17-氯甲基二甲基硅醚(II)的C3 位上的羰基不需要进一步保护,就可以在大位阻有机强碱的作用下,其本身含有的一个氯甲基与氰基发生分子内的环加成反应,再经水解就可以在氰基上引入一个含卤素原子的碳链[5]。经过这样一个中间态的环加成-水解反应,就可以避免21-H 的碘代反应,降低了生产成本,适合工业化生产。
由于化合物(I)与CDCS 的空间体积都较大,上述硅醚化反应很难发生。采用高效催化剂4-二甲氨基吡啶(DMAP)可解决这一问题。DMAP 能够在较温和的条件下让具有高位阻、低反应性的仲醇、叔醇、酰胺、胺等进行酰化、烷基化、醚化、酯化等多种类型的反应[6-8]。其催化活性要比吡啶、咪唑等高104~106[9]。
本文研究水分、pH、DMAP 用量及投料顺序等对反应的影响,并推测DMAP 参与化合物(I)的硅醚化反应历程。
1 实验部分
1.1 试剂与仪器
17β-氰基-17α-羟基雄甾-4-烯-3-酮,化学纯;氯甲基二甲基氯硅烷(CDCS)、4-二甲氨基吡啶(DMAP)、Et3N 等均为分析纯。
ANANCE Ⅲ-500 MHz 核磁共振波谱仪;DECAX-60000 LCQ Deca XP 液相色谱-离子阱质谱仪;Agilent 1100 高效液相色谱仪;PHSJ-5 实验室pH计。
1.2 实验方法
在氮气保护下,将DMAP 0.10 g(0.82 mol)用75 mL 二氯甲烷(DCM)溶解,加入Et3N 4. 78 mL(33.07 mmol)和CDCS 3.20 mL(24.29 mmol),降温至0 ℃,加入化合物(I)5. 18 g(16. 53 mmol),5 ℃保温反应2 h。加入1 mL 冰水淬灭,用10%盐酸水溶液调pH 至3,并补加25 mL 水,静止分层。水相用二氯甲烷2 ×15 mL 萃取2 次,合并有机相。依次用饱和NaHCO3水溶液和饱和NaCl 水溶液各洗1 次,无水硫酸钠干燥3 ~4 h。过滤,减压浓缩,得白色硅醚化物(II)。55 ℃真空烘料,得6.85 g 固体,收率98.70%。HPLC 检测纯度 为99. 07%,m.p.143.7 ~145.9 ℃(文献值:143 ~146 ℃)[10]。MS m/z=420.2[M]+;1H NMR(500 MHz,CDCl3)δ:0.38(s,6H,Si—CH3),0.97(s,3H,18—CH3),1.22(s,3H,19—CH3),2.88(m,2H,2—CH2),5.75(s,H,4—CH)。

2 结果与讨论
2.1 水分对目标产物收率的影响
2.1.1 反应体系中水分对目标产物收率的影响由表1 可知,反应体系中有水存在时,会对化合物(I)的硅醚化反应收率有显著的影响。反应初始时体系中的水分含量越多,对反应的影响就越大。这可能是由于初始时水分越多,则会有越多的CDCS遇水反应而失活,导致反应的初始速度大大降低。

表1 水分对反应的影响Table 1 Effects of moisture on the reaction
2.1.2 封闭体系水分随反应时间的变化 由表2可知,封闭体系,水分初始含量≤0. 03%,化合物(II)的收率随反应时间延长而增大,≥120 min 时,其收率基本不变。而且得到的硅醚化物(II)不会重新生产化合物(I)。

表2 反应时间对反应的影响Table 2 Effects of time on the reaction
2.1.3 敞开体系水分随反应时间的变化 由表3可知,敞开体系,水分会不断的进入到反应体系中,当CDCS 没消耗完之前,反应体系中无水;当CDCS消耗完全,随着水分的进入,生成的硅醚化物(II)又会重新生成化合物(I)。水分越多,对反应的影响就越大。

表3 反应时间和水分对反应的综合影响Table 3 Effects of time and water on the reaction
由上可知,封闭体系下,水分初始含量≤0.03%时,反应较易发生,水分是化合物(I)的硅醚化反应能否成功的关键。可能原因如下:一方面由于CDCS 较为活泼,遇水直接生成HCl 而失活;另一方面,水在催化剂作用下,与目标产物反应,重新生成化合物(I),反应机理如下。

2.2 后处理过程pH 对目标产物收率的影响
由表1 可知,DCM 和THF 均可以作为反应溶剂,并可以获得较好的收率。但是从溶剂回收难易及后处理考虑,选定以二氯甲烷(DCM)作为反应溶剂。初步推测水分使目标产物(II)重新生成化合物(I)的根本原因可能是由DMAP 的存在引起的。通过调节水相的pH 值来观察后续的处理过程中水分对目标产物(II)收率的影响,结果见表4。
由表4 可知,当水层pH >4 时,会有部分产物(II)重新生成化合物(I),pH 越大,产物(II)重新生成的化合物(I)就越多;当pH≤4 时,后续处理过程中水分对目标产物基本无影响。这可能是因为DMAP 是有机弱碱,只有在酸性环境中才能全部成盐,溶解在水中而完全除去。
由此可以推出:后处理过程中,如果没有DMAP,体系中即便有大量的水分存在,生成的目标产物也不会重新生成底物。即在水的存在下,DMAP 是使目标产物(II)重新分解为化合物(I)的关键。

表4 后处理过程中pH 对目标产物纯度的影响Table 4 Effects of pH of post-processing to the purity of the target product
2.3 催化剂DMAP 用量对化合物(I)硅醚化反应速率的影响
一般认为DMAP 是作为缚酸剂来参与反应,但当选用碱性较强的三乙胺代替DMAP 参与化合物(I)的硅醚化反应时,反应不会发生。由此可知,DMAP 在该反应中的主要作用并不是缚酸,而是以另一种形式参与反应。
2.3.1 投料顺序的影响 由表5 可知,除投料顺序不同,其它条件均相同时,相同的反应时间(10 min)内,将化合物(I)投入到DMAP 和CDCS 的混合液中时,化合物(I)的转化率要大于将CDCS 投入到化合物(I)和DMAP 的混合液中时化合物(I)的转化率,即反应速率前者比后者要快。原因可能是CDCS 与DMAP 首先生成(1∶1)络合物(1)(见图1)[11-13],该络合物的硅原子上有一个sp3d2杂化的空轨道,易接受化合物(I)中17-OH 氧原子上的孤对电子,从而促进了CDCS 与化合物(I)的反应。

表5 DMAP 用量对反应的影响Table 5 Effects of the consumption of DMAP on the reaction
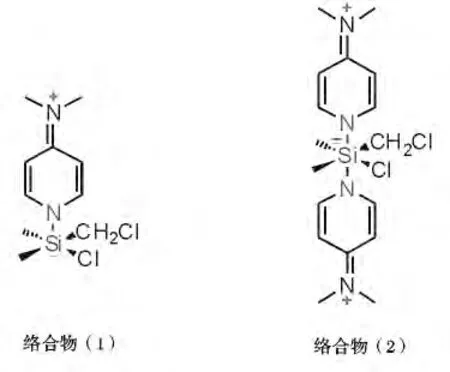
图1 CDCS 与DMAP 形成的络合物的结构式Fig.1 Structure of complexes of CDCS with DMAP
2.3.2 DMAP 用量的影响 由表5 可知,当投料顺序相同时,化合物(I)的转化率随DMAP 的变化如下。
当将化合物(I)投入到DMAP 与CDCS 的混合溶液中时,化合物(I)的反应速率将随着DMAP 用量的增大而加快。DMAP 用量(约1.65eq)到达一定值后,化合物(I)的反应速率达到一个最大值。此时,理论上CDCS 与DMAP 将全部生成络合物(1),CDCS 全部被活化,从而使化合物(I)的硅醚化反应速率达到最大。当DMAP 的量进一步增大时,将有部分CDCS 与两分子的DMAP 发生反应,生成络合物(2)。与络合物(1)相比,络合物(2)的硅原子上的两个sp3d2杂化轨道均被DMAP 吡啶环上N的孤对电子占据,又因化合物(I)的空间体积比DMAP 大得多,所以化合物(I)较难将DMAP 替换下来,从而导致化合物(I)的硅醚化反应速率随着DMAP 量的增大而有所降低。
当将CDCS 投入到DMAP 与化合物(I)的混合溶液中时,化合物(I)的反应速率随DMAP 量的增大而增大;但DMAP 用量到达一定值后,化合物(I)的转化率随DMAP 量的增多而基本不变。这可能是由于一开始DMAP 的量少,则主要与CDCS 生成络合物(1),化合物(I)的反应速率则随着络合物(1)含量的增多而加快;当DMAP 量足够多时,底物(I)与DMAP 在进攻络合物(1)上存在着竞争关系,再增加DMAP 的用量反应速率将不会有明显的提高。
由此可知,该反应的反应历程为:化合物(I)与CDCS 进行硅醚化反应时,首先是CDCS 与DMAP 生成络合物(1),使其活化,从而促使反应能够发生。DMAP 的用量为催化剂量时,便可以很大程度上提高该反应的速度。而且化合物(I)投入到DMAP 与CDCS 的混合溶液中时,反应较为有利。
2.4 反应机理
由上可知,化合物(I)的硅醚化反应可能经历了两个阶段:首先是一分子的DMAP 与一分子的CDCS 形成(1∶1)络合物(1)(图1);然后该络合物进攻化合物(I)的17 位羟基,再除去一分子的DMAP·HCl 盐,得到硅醚II。而DMAP·HCl 盐可以与碱性较强的三乙胺反应,将DMAP 释放出来,使DMAP 重新参与到反应中去。
硅原子属于第三周期元素,除s 及p 轨道外,还有5 个可供成键的空3d 轨道。当DMAP 与CDCS结合后,使得硅原子的杂化轨道由sp3杂化变为sp3d2杂化。由于络合物(1)的硅原子上含有一个sp3d2杂化空轨道,很容易接受化合物(I)的17-OH氧原子上的一对孤对电子,形成一个八面体结构,由于该八面体结构键角均为90°,空间位阻较大,同时氯原子又是一个较易离去的基团,所以该八面体结构很容易离去一分子HCl 和一分子DMAP,生成17β-氰基-17α-羟基雄甾-4-烯-3-酮-17-氯甲基二甲基硅醚(II)。可能的反应机理如下。

3 结论
(1)通过对水分、pH、DMAP 用量及投料顺序等的研究,确定了最优反应条件及较佳的投料顺序。氮气保护下,于0 ℃将化合物(I)加入到DMAP、Et3N、CDCS 的二氯甲烷混合溶液中,5 ℃保温反应2 h,冰水淬灭,分别用盐酸水溶液、碳酸氢钠溶液、食盐水各洗1 次,干燥、减压浓缩得白色硅醚化物(II),收率为98.70%,纯度为99.07%。
(2)DMAP 催化的化合物(I)与CDSC 的硅醚化反应的可能机理为:CDSC 先与DMAP 形成1∶1 的络合物,使其活化,进而与化合物(I)反应结合形成一个八面体络合物,该络合物在空间位阻的作用下,同时失去一分子HCl 和一分子DMAP,最终得到目标产物(II)。参考文献:
[1] Nitta I,Fujimori S,Ueno H.The synthesis of the corticoid side chain. I An improved method for the preparation of 17α-hydroxyprogesterone form androst-4-ene-3,17-dione[J].Bull Chem Soc Jpn,1985,58(3):978-980.
[2] Kataoka H,Watanabe K,Miyazaki K,et al. A short and efficient synthetic method for the corticosteroid side-chain from 17-keto steroids[J].Chem Lett,1990,19(9):1705-1708.
[3] Carruthers N I,Garshasb S,McPhail A T. Synthesis of corticoids from 9. alpha-hydroxyandrost-4-ene-3,17-dione[J].J Org Chem,1992,57(3):961-965.
[4] Gregory R J,Debiak K T. Corticoids from 17-oxosteroids[J].Tetrahedron Lett,1990,31(26):3669-3672.
[5] Livingston D A,Petre J E,Bergh C L.Intramolecular cyanohydrin elaboration. Construction of corticosteroids from 17-ketosteroids[J]. J Am Chem Soc,1990,112:6449-6450.
[6] Mermerian A H,Fu G C. Catalytic enantioselective synthesis of quaternary stereocenters via intermolecular C-acylation of silyl ketene acetals:Dual activation of the electrophile and the nucleophile[J]. J Am Chem Soc,2003,125:4050-4051.
[7] Neises B,Steglich W.Simple method for the esterification of carboxylic acids[J]. Angew Chem Int Ed,1978,17(7):522-524.
[8] Murugan R,Scriven E F V. Applications of dialkylaminopyridine catalysts in organic synthesis[J].Aldrichim Acta,2003,36:21-27.
[9] Yanchuk N I,Ivanets L N.Correlation of the catalytic activity of pyridines and N-pyridine oxides with their basicity[J].Russ J Gen Chem,2002,72(9):1453-1456.
[10] Livingston D A. 17β-cyano-9α,17α-dihydroxyandrost-4-en-3-one:US,4921638A[P].1990-05-01.
[11]Boudjouk P,Kloos S D,Kim B K,et al.An unexpected redistribution of trichlorosilane. Synthesis,structure and bonding of (N,N,N',N'tetraethylethylenediamine)dichlorosilane[J]. J Chem Soc Dalton Trans,1998(6):877-880.
[12]Fester G W,Wagler J,Brendler E,et al.Stable trichlorosilane-pyridine adducts[J].Eur J Inorg Chem,2008(32):5020-5023.
[13] Fester G W,Wagler J,Brendler E,et al.Octahedral HSiCl3and HSiCl2Me adducts with pyridines[J].J Am Chem Soc,2009,131:6855-6864.
猜你喜欢
能源化工(2021年2期)2021-12-30 18:31:06
昆明医科大学学报(2021年2期)2021-03-29 07:42:14
四川警察学院学报(2019年6期)2019-12-28 07:20:06
陕西中医(2018年6期)2018-08-29 00:43:34
文化产业(2016年6期)2016-10-19 19:13:47
中国塑料(2016年1期)2016-05-17 06:13:00
广东石油化工学院学报(2016年6期)2016-05-17 05:17:25
读写算·教研版(2016年8期)2016-05-07 11:52:08
中国塑料(2016年11期)2016-04-16 05:25:55
合成化学(2015年5期)2015-03-26 06:02:22
