
靶向乳腺癌的多西他赛壳聚糖纳米粒抑瘤作用研究
2015-04-14 07:47李庆
中国现代医学杂志 2015年3期
李庆
(长沙卫生职业学院,湖南 长沙410100)
多西他赛(docetaxel,DTX)为第二代紫杉烷类半合成的新型抗肿瘤药物[1],其作用机制是通过刺激导管素的聚合,促进微管双聚体装配成微管,并致使细胞增殖停止在有丝分裂静止期(G2/M)的阶段。目前已经成为激素难治性前列腺癌、乳腺癌及非小细胞肺癌等的一线治疗药物。由于多西他赛水溶性较差[2],目前临床上应用的唯一剂型是由40 mg/ml DTX和1 040mg/m l Tween 80组成的注射剂,临用前用13%的乙醇为专用溶剂复溶。由于Tween 80在体内会产生严重的变态反应,临床上需预先服用抗组胺剂和糖皮质激素,加之Tween 80会干扰DTX与血清白蛋白以浓度依赖的形式结合,改变药物的体内行为,使其呈非线性药物动力学[3]。
壳聚糖(chitosan,CS)作为优良的天然聚阳离子材料,具有良好的可生物降解、生物相容等性能,己被用于药物控释、靶向以及智能给药等多种药物载体的研究。肿瘤组织与正常组织在结构、基因表达及生物因子表达等方面存在差异,肿瘤细胞表面或其新生血管表面存在一系列特异表达或过度表达的抗原或受体,这些抗原或受体可以作为肿瘤靶向药物载体系统的结合靶点[4]。整合素作为细胞黏附分子家族,在肿瘤组织中广泛表达,其与细胞外基质配体结合特异性的精氨酸-甘氨酸-天冬氨酸(arginine-glycine-aspartic acid,RGD)序列[5],因此该特异性序列可作为肿瘤特异性靶点,介导靶向药物与肿瘤组织的识别、结合与相互作用。
本研究试图制备一种靶向乳腺癌组织的空间稳定性多西他赛壳聚糖纳米粒,以期改善传统药物剂型的不良影响,实现多西他赛的体内长循环,改善其在重要器官和肿瘤组织中的分布,减轻不良反应,同时提高其对肿瘤的治疗效果。
1 材料与方法
1.1 实验材料
1.1.1 主要试剂 壳聚糖和多西他赛购自美国Sigma公司;艾素购自江苏恒瑞医药股份有限公司;人乳腺癌细胞株MCF-7由中南大学细胞生物中心提供;氯仿、无水甲醇和乙酸乙酯等溶液均为分析纯。
1.1.2 主要仪器和耗材 SENCO-SHB(Ⅲ)水循环真空泵和RE-852旋转蒸发仪购自上海亚荣仪器公司;Ⅳ92-11超声细胞粉碎机购自宁波新芝生物科技股份有限公司;微脂粒挤压器为美国Avanti Polar Lipids公司产品;Ultracel YMl00超滤管为美国Millipore公司产品;高效液相色谱分析仪为日本岛津公司产品;二氧化碳培养箱为日本Sanyo公司产品;JEM-1200EXⅡ型透射电子显微镜为日本leol公司产品;Zeta Plus型Zeta电位及颗粒粒度分析仪为英国马尔文公司产品。
1.1.3 实验动物 SPF级BALB/c雌性裸鼠,4周龄,体重为(18±2)g。清洁级昆明小鼠,雌雄各半,4周龄,体重为(22±2)g。以上两种小鼠均购自中南大学动物实验学部,合格证编号分别为2007000502449和2007000506816。将乳腺癌细胞MCF-7培养至对数生长期,胰酶消化,然后用无血清培养液稀释成细胞悬液,取1×106个(0.1ml)细胞接种于BALB/c裸鼠右腋皮下。肿瘤大小按以下公式计算:肿瘤体积V(mm3)=a×b2/2,其中a为实体瘤最长径(mm),b为实体瘤最短径(mm)。
1.2 实验方法
1.2.1 RGD接枝壳聚糖形成RGD-CS[6]酰化反应偶联RGD与CS形成RGD-CS。称取RGD肽20.0mg,用1% HAc-NaAc缓冲液(pH 6.0)2m l溶解。加入EDC·HCl 100mg,NHS 50mg,在4℃下磁力搅拌,活化12 h。称取相对分子质量为50×103的CS 20mg溶于适量1%HAc溶液中,搅拌溶解。用1%NaOH溶液调节pH至6.0,得到黄色澄清溶液。在搅拌下,缓慢滴入活化的RGD溶液中。4℃下继续搅拌24 h。将反应液转移至透析袋中,用去离子水透析3 d,每12 h更换1次透析液。透析完毕后,产物冻干待用。
1.2.2 多西他赛壳聚糖纳米粒的制备 采用分散-交联法制备。精密称量多西他赛溶于1%醋酸溶液,超声溶解后,将壳聚糖细粉溶于其中配成一定浓度的壳聚糖溶液,静止除去气泡后用注射器缓缓滴入搅拌的混合油相中。分散一定时间后,加入一定量用戊二醛饱和的交联剂,固化。将产物分别用异丙醇、亚硫酸氢钠、石油醚洗涤3次,抽滤后,40℃干燥6 h,所得产物即多西他赛壳聚糖纳米粒(CS-DTX)。
同样条件下,采用RGD接枝壳聚糖(RGD-CS)与多西他赛制备靶向多西他赛壳聚糖粒(RGD-CS-DTX)。
1.2.3 检测多西他赛壳聚糖粒的包封率及物理性状
制备的多西他赛壳聚糖纳米粒经超滤(10 000 r/min,1 h)后,收集下清液,高效液相色谱法测定其中的多西他赛含量,记为ρ 游离。等容量壳聚糖经醋酸完全破膜后,测定其中的多西他赛含量,记为ρ 总。高效液相色谱法测定时,色谱柱为Diamond ODSC18柱(北京迪科马科技有限公司产品),流动相为甲醇:水(80:20),检测波长为227 nm,流速为1.0ml/min,进样量为20μl。在此色谱条件下,多西他赛的保留时间约为6.8min。包封率=(ρ 总-ρ游离)/ρ 总×100%。透射电子显微镜下观察纳米粒的形状特征,并用颗粒粒度分析仪测定其粒径分布。
1.2.4 检测多西他赛壳聚糖纳米粒的药物动力学和组织分布 荷瘤裸鼠模型的肿瘤大小生长至约100~200mm3时,分别单剂量尾静脉注射艾素、CS-DTX和RGD-CS-DTX(多西他赛剂量均为10 mg/kg),分别于注射后0.15、0.5、1、2、4、6、9和12 h(每个时间点各3只小鼠)时,摘除眼球取血,血液经抗凝处理,然后离心取血浆,于-80℃保存。药物注射后0.5、1、2、4、6和9 h这6个时间点,取小鼠的心、肝、脾、肺、肾及肿瘤组织,清洗称重,-80℃保存。取样结束后以颈椎脱臼法处死小鼠。血浆样品中加入无水甲醇,充分沉淀蛋白,离心取上清液,真空旋转蒸干,残留物用流动相溶解,高效液相色谱法检测多西他赛含量(方法同1.2.3)。组织样品加入匀浆液Tris-HCl(0.01mol/L)0.9ml,充分匀浆,匀浆液用乙酸乙酯萃取、离心后收集上层有机溶液,真空旋转蒸发,残留物用流动相溶解,高效液相色谱法检测多西他赛含量。然后分别将艾素、CS-DTX和RGD-CSDTX组血浆、组织的时间-药物浓度数据输入药代动力学3p87程序,以赤池信息准则(akaike information criterion,AIC)和拟合优度(r2)为判别指标,确定药物分布模型。根据各组织时间-多西他赛浓度数据,计算得出各组织的药物浓度-时间曲线下面积(area under curve,AUC),表示各组织中多西他赛累积总量。药物靶向效率(targeting efficiency,t)=AUC组织/AUC血,肿瘤组织相对摄取量=AUC肿瘤/AUC组织。
1.2.5不同剂型多西他赛的毒性实验 昆明小鼠在适应实验室环境1周后,给予正常饮食,并随机分为4组。24 h内分别尾静脉注射艾素、CS-DTX和RGD-CS-DTX,同时以注射等体积的0.9%氯化钠溶液作为对照组。观察期至注射后第7天,每天测量小鼠体重,观察结束后与给药前体重比较,以体重降低≤10%的最高剂量作为最大耐受量,体重降低>20%的最高剂量作为致死剂量。预实验中,每组2只小鼠,起始剂量为10mg/kg,若小鼠观察期内未出现死亡或未达到体重降低10%,则增加注射剂量。艾素递增剂量为5mg/kg,脂质体制剂递增剂量为10mg/kg,直到小鼠出现体重降低10%,且未出现致死剂量反应时,则以每组10只小鼠在该剂量下重复实验,确定最大耐受量。
1.2.6 不同剂型多西他赛的体内抑瘤作用 荷瘤裸鼠模型建立7 d后(此时记为d 1),随机分为6组,每组5只,分别尾静脉给药。组1:0.9%氯化钠溶液等体积注射,作为阴性对照;组2:艾素10mg/(kg·d)(共60mg/kg);组3:CS-DTX 10mg/(kg·d),d1~6(共60 mg/kg);组4:RGD-CS-DTX 5mg/kg,d1~6(共30mg/kg);组5:RGD-CS-DTX间隔给药10mg/(kg·d),d1,d3,d5(共30mg/kg);组6:RGD-CS-DTX 10mg/(kg·d),d1~6(共60mg/kg)。d7起密切观察荷瘤小鼠的一般状况,每周2次测量体重和肿瘤体积。待肿瘤大小超过2 000mm3,达到小鼠最大荷瘤状态时结束观察,小鼠称重并摘除眼球取血,颈椎脱臼法处死,剥离肿瘤,记录瘤重。肿瘤抑制率计算公式:抑制率=(1-给药组瘤重/对照组瘤重)×100%。
1.3 统计学方法
采用SPSS 18.0软件对实验数据进行统计学分析。定量数据以均数±标准差(±s)表示,不同组别问比较采用单因素方差分析和LSD检验,以P<0.05为差异有统计学意义。
2 结果
2.1 多西他赛壳聚糖纳米粒的包封率及物理性状
超滤法测得多西他赛纳米脂质体的包封率为(96.84±3.74)%。透射电子显微镜下观察可见,CS-DTX及RGD-CS-DTX均呈圆形囊泡样结构,大小较一致,分散均匀(图1)。粒径分布检测结果显示,CS-DTX及RGD-CS-DTX的平均粒径分别为(84.2±22.6)nm和(95.2±28.5)nm。见图2。
2.2 多西他赛回收率
高效液相色谱法检测后,以峰面积A对质量浓度C(μg/ml)进行线性回归,结果显示浓度C在0.192~26μg/m l内峰面积A呈良好线性关系,回归方程为A=84826C+4673,r2=0.9999。精密配制质量浓度为0.195、1.56和12.5μg/m l的多西他赛标准溶液,于1 d内重复测定3次,得日内精密度的相对标准偏差(relative standard deviation,RSD)为(3.12±1.34)%(n=9);于数天内重复测定3次,得日间精密度RSD为(3.62±1.27)%(n=9)。用多西他赛浓度测定法计算回收率:精密量取空白脂质体溶液1.0ml,加入适量的多西他赛对照品,使得脂质体中多西他赛质量浓度为0.195、1.56和12.5μg/ml,依法操作测定多西他赛浓度,多西他赛平均回收率为(99.16±3.87)%(n=9)。
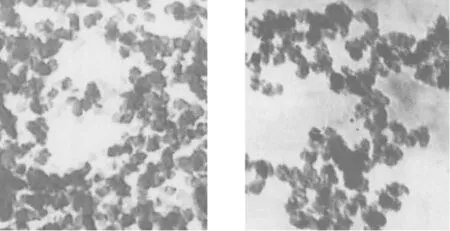
图1 CS-DTX及RGD-CS-DTX的透射电子显微镜下观察图(醋酸铀染色,×30 000)
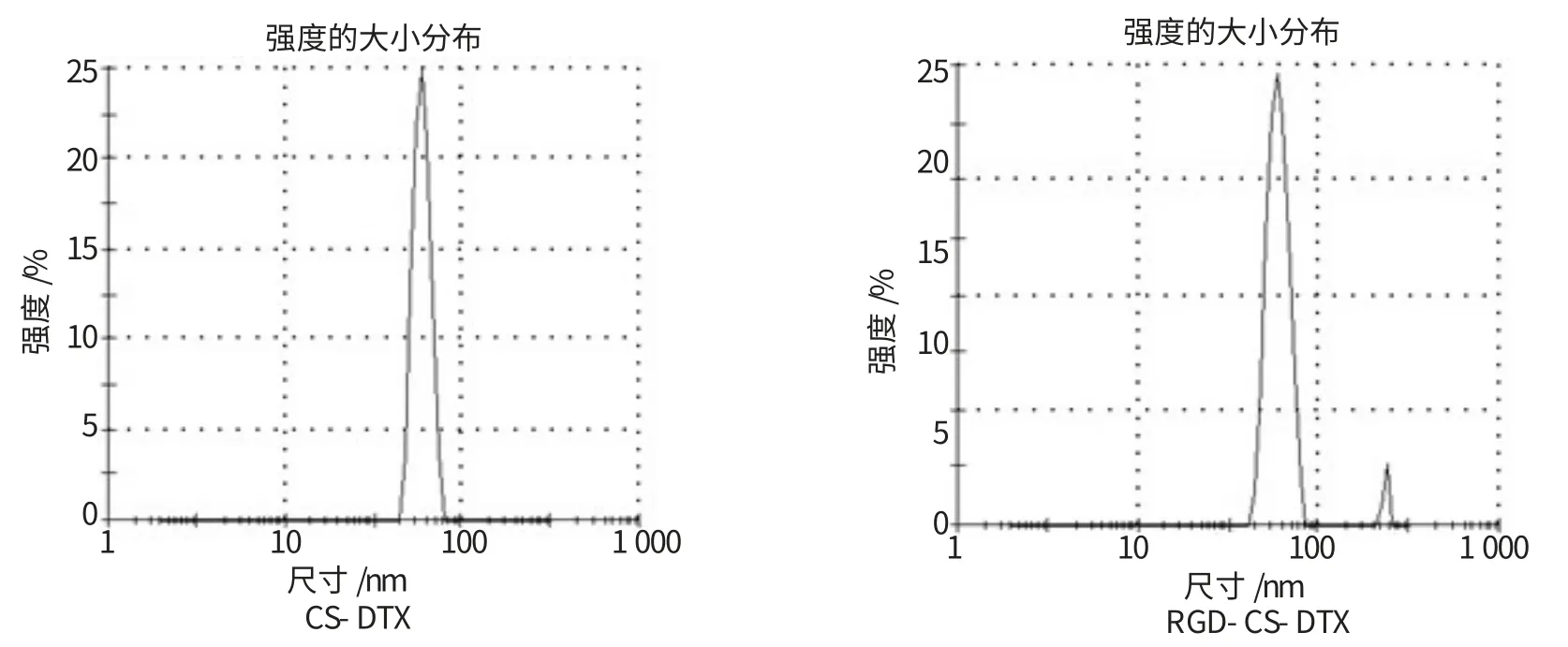
图2 CS-DTX及RGD-CS-DTX的粒径分布图
2.3 药物动力学和组织分布
药物动力学分析以1/c2为权重,对数据进行模型嵌合,结果显示艾素和多西他赛脂质体均呈二室模型。相比艾素,壳聚糖剂型的半衰期(half-lifetime,t1/2)明显延长,清除率降低,AUC0-∝增高,平均滞留时间延长(P均<0.05)。提示药物循环时间延长,清除减慢,进入血液循环的总量增加,体内达到长循环,从而有更多的药物能有效地从血循环分布到靶器官。RGD-CS-DTX与CS-DTX相比,各药物动力学参数无明显差异,提示血管靶向多肽复合物的加入未影响药物在体内的药物动力学(表1)。
组织分布检测结果显示,艾素组各组织中多西他赛含量随时间延长而迅速降低,6 h后仅有微量浓度检出;而CS-DTX及RGD-CS-DTX组各组织中多西他赛清除较艾素组缓慢,9 h后仍测出较高浓度,其中肝、脾及肿瘤组织中多西他赛含量较高,尤其在肿瘤组织中,多西他赛含量可较长时间地保持高水平,清除较慢。RGD-CS-DTX经尾静脉注射后,肿瘤组织靶向效率提高,RGD-CS-DTX、CS-DTX及艾素组Te肿瘤/血分别为2.42、1.07和0.35(P均<0.05,表2),提示肿瘤主动靶向多西他赛纳米脂质体能增强药物的靶向能力,使药物更多地分布于肿瘤组织,而其他正常器官则分布相对较少;肿瘤组织相对于心、肺、肾的摄取率明显增高,RGD-CS-DTX组相比CS-DTX和艾素组的AUC肿瘤/AUC心、AUC肿瘤/AUC肺和AUC肿瘤/AUC肾均有明显提高(均P<0.05,表2)。

表1 不同剂型多西他赛(10mg/kg)在静脉注射后的体内药物动力学参数

表2 不同剂型的多西他赛在荷瘤裸鼠模型体内分布的靶向效率和肿瘤组织相对摄取比
2.4 最大耐受量
给予昆明小鼠不同剂型的多西他赛后发现,艾素给药组小鼠先出现短暂的兴奋,随后出现卧伏和运动减少,10min后恢复正常;壳聚糖给药组小鼠未出现异常反应。0.9%氯化钠溶液对照组小鼠无死亡,体重无明显降低;艾素、CS-DTX和RGD-CS-DTX的最大耐受量分别为35、220和220mg/kg,且雌雄小鼠无差别。
2.5 体内抑瘤作用
荷瘤小鼠给予不同剂型、不同剂量和不同给药间期的多西他赛7 d后(记为d 7),密切观察肿瘤生长情况,绘制肿瘤生长曲线(图3A)。d 7时对照组的瘤体大小均超过2 000mm3,因此结束观察,摘除肿瘤,测瘤重(图3B),计算抑瘤率。计算结果显示,给予艾素60mg/kg治疗时,脂质体组的抑瘤率>艾素组,而RGD-CS-DTX的抑瘤作用优于CS-DTX(P<0.05);当RGD-CS-DTX低剂量给药或延长给药间期时,其抑瘤作用也优于CS-DTX(P<0.05,表3)。

表3 艾素、CS-DTX及RGD-CS-DTX的抑瘤率

图3 给予不同剂型的多西他赛治疗后荷瘤裸鼠体内肿瘤的生长曲线和剥离后照片图
3 讨论
目前,化疗已成为肿瘤治疗的重要手段之一。然而,化疗药物由于缺乏作用的选择性而常损伤正常器官,引起严重的全身不良反应。主动靶向的纳米壳聚糖因具有下列特点而成为化疗药物的良好载体:①壳聚糖体具有生物相容性,毒性较小;②纳米粒径的壳聚糖易透过血液屏障到达靶器官,且比表面积大,能在其表面偶联大量的效应分子;③聚乙二醇(polyethylene glyol,PEG)修饰的长循环脂质体被肝脾吞噬明显减少,在血液中有足够的循环半衰期;④靶向配体与壳聚糖相连,不影响脂质体内包封药物的结构和生物学特性,且载药量大;⑤通过配体—受体作用,特异靶向肿瘤组织,使药物浓聚于靶器官,增强靶细胞对药物的结合,减少其他器官的分布[7-8]。
整合素介导细胞与细胞外基质的相互作用,在肿瘤细胞生长、迁移及肿瘤新生血管生成中发挥重要作用。整合素为跨膜异二聚体糖蛋白,能与细胞外基质配体结合,其识别位点包含特异性的RGD序列。含有RGD序列的多肽配体,能竞争结合位点,抑制整合素功能,作为主动靶点介导药物与肿瘤组织的特异性识别和结合。壳聚糖作为一种天然阳离子聚合物的药物载体,具有细胞毒性低、生物相容性好及基因免疫性低等优点。但其细胞摄取效率较低,生物靶向性较差,因而也限制了它在临床上的应用。本研究将壳聚糖和RGD肽通过化学键连接,以纳米粒子形式包载多西他赛,其肿瘤细胞的靶向效率明显高于单纯的壳聚糖。表明RGD肽对细胞表面的靶向结合作用能增强壳聚糖输送药物进入细胞内部发挥生物学作用。壳聚糖-RGD纳米粒同时具有壳聚糖和RGD肽的优点,显示出了很强的应用价值。
此外,本实验制备的多西他赛壳聚糖纳米粒CS-DTX和RGD-CS-DTX,在血浆中循环半衰期明显延长,肿瘤组织浓聚明显增加,未见明显的肝、脾摄取,且心、肺、肾摄取也明显降低,因此其对重要器官的毒性降低,药物最大耐受量明显提高。推测其主要原因,可能如下:①制备的PEG修饰的多西他赛壳聚糖纳米粒<100 nm,减少了血浆调理素、网状内皮细胞与脂质体的相互作用,肝、脾摄取减少;②多西他赛壳聚糖纳米粒的相对分子质量较大,不易通过肾毛细血管滤过,肾脏代谢降低;③肿瘤组织的增强渗透和滞留作用(enhanced permeation and retention,EPR)使多西他赛壳聚糖纳米粒易于聚集在肿瘤组织中,CS-DTX与RGD-CS-DTX的肿瘤摄取量均高于艾素;④连接RGD肽的RGD-CS-DTX特异性结合至肿瘤组织表达的整合素受体,肿瘤摄取百分比约为CS-DTX和泰素的2和4倍。
本研究中体内疗效观察发现,艾素、CS-DTX和RGD-CS-DTX各给药组均有抑瘤作用,给药后一段时间内艾素组的肿瘤生长逐渐与对照组呈相同增长趋势,在观察结束时其瘤重与对照组无显著差异;相反,各脂质体给药组的肿瘤生长保持相对缓慢。脂质体组内分析结果显示,RGD-CS-DTX予以高、低剂量或间隔给药,其抑瘤作用均优于CS-DTX(P<0.05)。组织分布检测结果显示,RGD-CS-DTX的肿瘤组织靶向效率约为CS-DTX的2倍(P<0.05);予以CS-DTX剂量1/2的RGD-CS-DTX时,后者的抑瘤作用仍优于前者,这与组织分布靶向效率结果一致。推测RGD-CS-DTX通过受体一配体结合作用,介导靶细胞结合载药脂质体,药物浓聚于肿瘤组织,从而达到更强的抑瘤作用。药代动力学分析结果显示,RGD-CS-DTX及CS-DTX的消除半衰期t1/2β明显长于艾素,因此延长其给药间隔时药物仍能维持一定的抑瘤有效浓度,RGD-CS-DTX间隔给药组的疗效显著优于艾素及CS-DTX组。
综上所述,本研究制备的乳腺癌靶向的多西他赛壳聚糖纳米粒,可有效改善多西他赛药物的体内分布,提高其体内安全性和疗效,有望成为一种有效的新型抗肿瘤药物载体。
[1]ALKEN S,KELLY CM.Benefit risk assessmentand updateon the use of docetaxel in themanagementof breast cancer[J].CancerManag Res,2013,5(4):357-365.
[2]ZHANG H,DOU J,ZHAIY,et al.Advances in the formulations of non-injectionadministrationofdocetaxel[J].JDrug Target,2014,22(2):87-94.
[3]TANQ,LIUX,FUX,etal.Currentdevelopmentinnanoformu-lationsof docetaxel[J].ExpertOpin Drug Deliv,2012,9(8):975-990.
[4]LUO Y,WANG Q.Recent development of chitosan-based polyelectrolyte complexes with natural polysaccharides for drug delivery[J].Int JBiolMacromol,2014,64(8):353-367.
[5]IYER AK,SINGH A,GANTA S,et al.Role of integrated cancer nanomedicine in overcoming drug resistance[J].Adv Drug Deliv Rev,2013,65(13-14):1784-1802.
[6]LIUWQ,MARTINEZ JA,DURAND J,etal.RGD-mediated adhesive interactions are important for peripheral axon outgrowth in vivo[J].NeurobiolDis,2009,34(1):11-22.
[7]SHUKLA SK,MISHRA AK,AROTIBA OA,et al.Chitosan-based nanomaterials:a state-of-the-art review[J].Int JBiolMacromol,2013,59(7):46-58.
[8]HU L,SUN Y,WU Y.Advances in chitosan-based drug delivery vehicles[J].Nanoscale,2013,5(8):3103-3111.
猜你喜欢
河北科技师范学院学报(2022年2期)2022-08-26
昆明医科大学学报(2022年3期)2022-04-19
当代水产(2021年3期)2021-07-20
河北科技师范学院学报(2021年1期)2021-05-10
视界观·上半月(2020年2期)2020-04-20
中成药(2018年2期)2018-05-09
中成药(2018年2期)2018-05-09
中成药(2017年12期)2018-01-19
美与时代·城市版(2018年10期)2018-01-16
中成药(2017年3期)2017-05-17
