
CRISPR/Cas9技术在HepG2基因组中进行定点基因突变的探索
2015-04-13 11:13肖利佳龙寿斌罗历沈化清郝建华
海南医学 2015年1期
肖利佳,龙寿斌,罗历,沈化清,郝建华
(广东医学院附属南山医院检验科,广东深圳518052)
CRISPR/Cas9技术在HepG2基因组中进行定点基因突变的探索
肖利佳,龙寿斌,罗历,沈化清,郝建华
(广东医学院附属南山医院检验科,广东深圳518052)
目的探索利用CRISPR/Cas9技术在细胞株HepG2基因组中进行定点突变。方法设计并构建靶向核受体LRH-1和ERRα基因组序列的gRNA质粒。将构建好的gRNA质粒和hCas9质粒转染HepG2细胞后,PCR扩增HepG2基因组中LRH-1和ERRα基因的突变位点,并用SURVEYOR法检测突变情况。结果靶向核受体LRH-1和ERRα基因组序列的gRNA质粒构建成功。HepG2细胞经gRNA-LRH-1/gRNA-ERRα和hCas9转染后,针对LRH-1和ERRα的突变位点基因组序列的PCR产物经SURVEYOR法检测结果显示出现两条与预计大小相符的电泳条带。结论在HepG2细胞中成功利用CRISPR/Cas9技术介导的基因组编辑对LRH-1和ERRα特定基因组序列产生突变,为构建其细胞株敲除模型奠定了基础。
CRISPR;HepG2;基因组编辑;定点突变
由依赖于规律成簇的间隔短回文重复(Clustered regularly interspaced short palindromic repeats,CRISPR)和Cas蛋白(CRISPR associated proteins)构成的CRISPR/Cas9系统是一类广泛存在于细菌和古细菌体内的免疫防御机制,可用来对抗入侵的病毒及外源DNA[1-3]。此系统中,crRNA(CRISPR-derived RNA)与tracrRNA(Trans-activating RNA)结合形成tracrRNA/ crRNA复合物,此复合物能引导核酸内切酶Cas9在与crRNA碱基配对的靶序列剪切双链DNA。随后研究证实通过异位表达CRISPR/Cas9系统中的关键因子,同样可以在哺乳动物基因组诱发双链DNA断裂[4]。哺乳动物基因组中的双链DNA断裂主要是通过随机插入或删除的非同源重组的末端修饰(Non-homologous end joining,NHEJ)进行修复,因此可以通过此种方法在哺乳动物基因组中诱发定点突变。以往研究证实将crRNA和tracrRNA融合在同一个质粒上表达,同样可以引导Cas9特异性地切断靶DNA序列[5-6]。因其快速、简便,该系统已被广泛应用于定点基因编辑和基因敲除模型的建立[7]。目前,对于利用CRISPR/Cas9系统在稳定癌细胞系中构建基因敲除模型的报道较少,因此我们拟利用CRISPR/ Cas9系统在肝癌细胞系HepG2基因组中进行定点突变,为探索在体外稳定肝癌细胞株HepG2中建立特定基因敲除模型提供参考依据。
1 材料与方法
1.1 材料CRISPR/Cas9系统中的gRNA和hCas9质粒购自addgene Church实验室。人肝癌细胞株HepG2购自ATCC,为本实验室保存,培养于含有10%FBS的DMEM培养基中。AflII限制性核酸内切酶、T7核酸内切酶Ⅰ(T7 EndonucleaseⅠ,T7EⅠ)和Gibson Assembly®Cloning Kit购于NEB公司(New England Biolabs),质粒提取试剂盒和PCR产物纯化试剂盒购自Qiagen公司,高保真DNA聚合酶KAPA HiFi HotStart Polymerase购自Kapa Biosystem公司。寡聚核苷酸序列由Tech Dragon公司合成,PCR引物由Invitrogen公司合成,DNA测序由华大基因公司完成。
1.2 方法
1.2.1 寡聚核苷酸的设计及合成由于gRNA载体中crRNA和tracrRNA融合体的转录是由U6启动子控制,并且CRISPR/Cas9系统中crRNA对20个碱基靶序列DNA识别时要求靶序列后紧跟一个-NGG(N代表ATGC中任意一个碱基)的PAM(Protospacer-adjacent motif)序列。因此针对核受体LRH-1和ERRα的基因组序列,我们分别设计GTAAGGGCCGACCGAATGCG(TGG)和GCAGAAGTACAAGCGGCGGC(CGG)作为其靶序列,相应的寡聚核苷酸序列由Tech Dragon公司负责合成。
1.2.2 gRNA-LRH-1和gRNA-ERRα质粒的构建将合成的0.1 nmol寡聚核苷酸序列正链和负链分别加入KAPA HiFi HotStart Polymerase反应体系中,按照95℃2 min,98℃20 s、65℃15 s、72℃15 s共 30个循环,最后72℃5 min充分延伸。取5µl退火延伸产物进行2%的琼脂糖凝胶电泳,随后对双链DNA延伸产物进行纯化。gRNA空载体经AflII限制性核酸内切酶消化后,将酶切产物和寡聚核苷酸退火延伸产物经纯化定量。于10µl的Gibson Assembly mix中加入100 ng经酶切纯化后的双链DNA产物和300 ng的gRNA空载体,用蒸馏水补齐20µl,于50℃孵育60 min将双链DNA产物连接到gRNA空载体上。随后将连接产物转化DH5α感受态细胞,铺板培养后选取单克隆扩大培养,提取质粒后交由华大基因公司测序验证。
1.2.3 细胞转染转染前一天将培养于含有10%FBS的DMEM培养基中的HepG2细胞经胰酶消化后重悬,并重新种入6空板内,第2天待HepG2细胞生长处于对数生长期时进行细胞转染实验。采用Lipo 2000脂质体介导的共转染法:首先形成Lipo/ gRNA-LRH-1/gRNA-ERRα和hCas9复合物,室温孵育30 min后将此复合物加入细胞培养液中,培养6 h后更换成含10%血清的培养基继续培养24 h。
1.2.4 基因组DNA的提取及突变的SURVEYOR验证HepG2细胞经Lipo/gRNA-LRH-1/ gRNA-ERRα和hCas9复合物转染24 h后,收集细胞加入细胞裂解液于37℃孵育45 min,随后加入200µl氯仿混匀,经离心分离后按1:1体积加入异丙醇,待白色絮状物形成后离心收集DNA沉淀,用70%的乙醇洗涤两次以后加入消毒蒸馏水溶解DNA并测定浓度。随后利用PCR技术扩增出突变位点,LRH-1突变位点引物上游5'-TCTCCTCCATCCACAAAAGG-3';下游5'-CTCGGGTGAGCTGGTATAGG-3'。ERRα突变位点引物上游5'-GCCTCCAACGAGTGTGAGAT-3';下游5'-GTGGCCTTCCTCAGTAGCAG-3'。PCR产物经胶回收纯化,在T7EⅠ缓冲液中先95℃5 min充分变性,然后按照1℃/min缓慢退火,待退火完成后加入T7EⅠ,消化产物随后进行1%的琼脂糖凝胶电泳检测。
2 结果
2.1 寡聚核苷酸的设计、合成及退火延伸根据CRISPR/Cas9系统对目的DNA序列识别的规律,我们设计了针对核受体LRH-1和ERRα基因组序列的寡聚核苷酸序列,其中黄色背景部分为反向互补序列,也是靶向目的DNA的识别序列(见图1)。将合成的0.1 nmol寡聚核苷酸序列正链和负链经退火延伸后,5µl退火延伸产物进行2%的琼脂糖凝胶电泳显示,针对LRH-1和ERRα的寡聚核苷酸序列退火产物在100 kb左右均出现明显的条带,与预计片段大小相符,且无其他非特异性片段,提示退火延伸成功(见图2)。
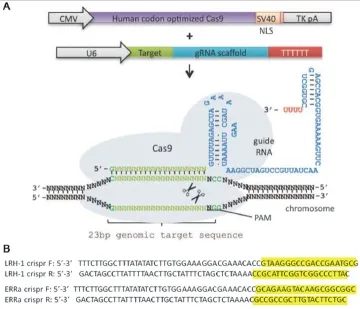
图1 CRISPR/Cas9系统和构建gRNA-LRH-1和gRNA-ERRα的寡聚核苷酸序列
2.2 寡聚核苷酸退火产物与gRNA载体的连接和验证gRNA空载体经AflII限制性核酸内切酶消化,酶切产物和寡聚核苷酸退火延伸产物经纯化定量后利用Gibson Assembly®Cloning Kit进行连接,连接产物转化DH5α感受态细胞,铺板培养后分别选取单克隆扩大培养,提取质粒后送华大基因公司测序分析,其测序结果与合成的寡聚核苷酸序列比对后完全一致,证实gRNA-LRH-1和gRNA-ERRα重组质粒载体构建成功(见图3)。

图2 寡聚核苷酸退火产物的电泳分析
2.3 基因组DNA的提取及突变的SURVEYOR验证HepG2细胞经Lipo/gRNA-LRH-1/gRNA-ERRα和hCas9复合物转染24 h后提取全基因组DNA,经定量后以基因组DNA为模板,以分别针对LRH-1和ERRα基因组突变位点的引物进行PCR反应,扩增产物经胶回收纯化后用T7EⅠ消化,随后对PCR产物和消化产物进行1%的琼脂糖凝胶电泳检测,电泳结果显示在对照组和实验组均能检测到与预计大小相符的目的条带。经T7EⅠ消化后,在转染gRNA-LRH-1/ hCas9组的电泳结果显示出现两条额外的358 bp和281 bp的片段,转染gRNA-ERRα/hCas9组出现两条额外的366 bp和316 bp的片段,与预计大小相符(见图4)。该结果说明部分HepG2细胞的基因组序列因为产生了由CRISPR/Cas9系统诱发的随机插入或删除的非同源重组的末端修饰,从而导致基因组突变而在PCR产物经缓慢退火时不能完全配对(Mismatch),因此能被T7EⅠ识别从而切断成两条小的DNA片段,证实CRISPR/Cas9系统成功在HepG2细胞基因组上导致定点突变。

图3 gRNA-LRH-1和gRNA-ERRα重组质粒载体的测序验证

图4 HepG2基因组DNA突变的SURVEYOR验证
3 讨论
CRISPR/Cas9技术是继锌指核酸酶(Zinc-finger nucleases,ZFNs)和转录激活因子样效应物核酸酶(Transcription activator-like effector nucleases,TALENs)技术后可用于定点基因组编辑并取得快速发展的新兴技术,且有效率高、速度快及简单经济的特点,在构建基因敲除动物模型的应用前景将非常广阔[8]。目前已有多个研究小组利用CRISPR/Cas9技术构建基因敲除小鼠并获得成功,其中来自剑桥大学的Rudolf Jaenisch课题组利用该技术在胚胎干细胞中同时敲除多达5个基因,并且通过受精卵注射,成功构建同时敲除两个基因的基因敲除小鼠[9]。尽管CRISPR/ Cas9技术近几年在构建基因敲除动物模型的应用取得了可喜的成绩,但是目前利用该技术在稳定癌细胞系中构建基因敲除模型的报道较少。
核受体是一类转录因子超家族,其通过受体依赖性或者非受体依耐性的方式调控下游基因表达,不仅在新陈代谢、生殖、发育等生理过程中起着重要的作用,其同样参与包括肝癌在内的多种癌症的发生和发展[10]。因此本研究选择两个重要核受体LRH-1和ERRα基因组序列作为靶序列,拟利用CRISPR/Cas9技术在肝癌细胞株HepG2基因组中对特定基因序列定点突变进行探索。根据CRISPR/Cas9系统中crRNA识别20个碱基的靶序列DNA时要求靶序列后紧跟一个PAM(NGG)序列的原则,我们首先设计合成了靶向LRH-1和ERRα基因组序列的寡聚核苷酸序列,并运用Gibson Assembly®Cloning Kit将退火延伸的寡聚核苷酸序列产物连接到gRNA载体,测序验证成功后转染HepG2细胞,并对转染后的HepG2基因组靶序列进行SURVEYOR实验以验证突变的产生。结果显示靶向LRH-1和ERRα基因组序列的CRISPR/Cas9系统在HepG2细胞中均导致了特定位点的基因突变。同时结果提示只有一部分HepG2细胞中产生了基因突变,这是因为将CRISPR/Cas9系统导入细胞有一定的转染效率,并且CRISPR/Cas9系统对靶基因的剪切和细胞的修复过程是一个动态的过程,另外癌细胞株中一部分基因存在基因组上拷贝数的扩增,这些原因使得不能利用CRISPR/Cas9系统针对大量细胞进行细胞转染从而获得基因敲除细胞株。若要得到完全敲除LRH-1和ERRα基因的细胞株则需要进行单克隆的筛选和鉴定。
本研究的前期结果证实了利用CRISPR/Cas9系统在肝癌细胞株HepG2基因组中对核受体LRH-1和ERRα基因组序列进行定点突变,为在HepG2细胞中建立LRH-1和ERRα基因敲除细胞模型奠定了基础。
[1]Wiedenheft B,Sternberg SH,Doudna JA.RNA-guided genetic silencing systems in bacteria and archaea[J].Nature,2012,482 (7385):331-338.
[2]Bhaya D,Davison M,Barrangou R.CRISPR-Cas systems in bacteria and archaea:versatile small RNAs for adaptive defense and regulation[J].Annu Rev Genet,2011,45:273-297.
[3]Terns MP,Terns RM.CRISPR-based adaptive immune systems[J]. Curr Opin Microbiol,2011,14(3):321-327.
[4]Cong L,Ran FA,Cox D,et al.Multiplex genome engineering using CRISPR/Cas systems[J].Science,2013,339(6121):819-823.
[5]Jinek M,Chylinski K,Fonfara I,et al.A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity [J].Science,2012,337(6096):816-821.
[6]Mali P,Yang L,Esvelt KM,et al.RNA-guided human genome engineering via Cas9[J].Science,2013,339(6121):823-826.
[7]Zhang F,Wen Y,Guo X.CRISPR/Cas9 for genome editing:progress,implications and challenges[J].Hum Mol Genet,2014,23 (R1):R40-46.
[8]Wilkinson R,Wiedenheft B.A CRISPR method for genome engineering[J].F1000Prime Rep,2014,6:3.
[9]Wang H,Yang H,Shivalila CS,et al.One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering[J].Cell,2013,153(4):910-918.
[10]Francis GA,Fayard E,Picard F,et al.Nuclear receptors and the control of metabolism[J].Annu Rev Physiol,2003,65:261-311.
Exploration of site-directed mutagenesis mediated by CRISPR/Cas9 system in HepG2.
XIAO Li-jia,LONG Shou-bin,LUO Li,SHEN Hua-qing,HAO Jian-hua.Department of Clinical Laboratory,Nanshan Affiliated Hospital of Guangdong Medical College,Shenzhen 518052,Guangdong,CHINA
ObjectiveTo explore whether CRISPR/Cas9 system could be effective in hepatocarcinoma cell line HepG2.MethodsgRNA plasmids targeting nuclear receptor LRH-1 and ERRα were designed and constructed.After verification by sequencing,gRNA-LRH-1/gRNA-ERR α and hCas9 plasmids were transfected in HepG2 cells respectively.Then mutation sites were amplified by PCR and mutation rates were evaluated by SURVEYOR assay.ResultsgRNA-LRH-1 and gRNA-ERRα recombination plasmids targeting nuclear receptor LRH-1 and ERRα were successfully established.After transfecting HepG2 with gRNA-LRH-1/gRNA-ERRα and hCas9,the mutation sites were amplified by PCR and subjected to SURVEYOR assay.The electrophoresis results showed that two additional bands with expected size were found in cells transfected with gRNA-LRH-1/gRNA-ERRα and hCAS9.ConclusionThe site-directed mutagenesis of LRH-1 and ERRα gene locus were successfully created by genome editing mediated by CRISPR/Cas9 in HepG2,which provides the basis for the generation of HepG2-LRH-1/ERRα knockout cell model.
CRISPR;HepG2;Genome editing;Site-directed mutagenesis
R37
A
1003—6350(2015)01—0007—04
10.3969/j.issn.1003-6350.2015.01.0003
2014-07-03)
深圳市卫生计生系统科研项目(编号:201402135)
郝建华。E-mail:haojmail@aliyun.com
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
中学生数理化(高中版.高考数学)(2022年4期)2022-05-25
今日农业(2021年21期)2021-11-26
新世纪智能(教师)(2021年2期)2021-11-05
教育周报·教育论坛(2021年21期)2021-04-14
中国饲料(2019年19期)2019-03-25
中成药(2018年3期)2018-05-07
医学研究杂志(2015年11期)2015-06-10
