
负载方式对Ag/Co3O4催化剂催化氧化甲醛活性的影响
2015-04-10 03:01张江浩王亚飞张长斌
化学工业与工程 2015年3期
张江浩,王亚飞,张长斌,贺 泓
(中国科学院生态环境研究中心,环境模拟与污染控制国家重点联合实验室,北京 100085)
甲醛是我国室内环境中最严重的污染物,多种室内装修材料都可能向室内释放甲醛。当人体暴露于甲醛浓度过高的污染气体中时,视甲醛浓度和暴露时间的不同,可能引起相应的不良反应或疾病。轻则刺激眼、鼻、喉等器官,重则引起咳嗽、疲劳等严重的过敏反应,甚至诱发组织癌变[1-2]。根据最新的研究结论,甲醛还会损害人体的神经系统和心血管系统[3]。因此,我国制定的室内空气质量标准(GB/T 18883-2002)中规定室内空气中甲醛浓度限值为0.1 mg/m3(小时均值)[4]。为了改善空气质量,降低人们的健康风险,提高人们的生活水平,研究高效去除室内甲醛污染的方法已经刻不容缓。
在众多去除甲醛的技术手段中,催化氧化法以其操作简单、无二次污染等优点成为最具前景的去除室内甲醛的方法。催化氧化技术的核心为催化剂,目前的催化剂主要分为贵金属催化剂和非贵金属催化剂,其中以Pt、Au和Pd为代表的贵金属催化剂[5-8]展示了极为优良的催化活性,即使在高空速条件下,也能够将高浓度的甲醛在室温下完全氧化为二氧化碳和水。但是,贵金属催化剂价格昂贵的缺点成为其广泛应用的最大障碍,与此同时Ag基催化剂以其较低的价格,较高的活性得到越来越多的关注。Qu等[9]采用后嫁接的方法制备的Ag/SBA-15能够在100 ℃将甲醛完全去除;Shi等[10]制备的Ag/MnOx-CeO2完全氧化甲醛的温度降至80 ℃。
甲醛催化氧化的重要步骤之一为甲酸分解形成的一氧化碳的氧化[11],在众多催化氧化一氧化碳的催化剂中,Co3O4表现出极高的活性,形貌的控制可以使Co3O4即使在有水汽存在的条件下,仍然能够在-77 ℃将CO完全氧化[12]。因此,在特定形貌下,Ag与Co的协同(负载型Ag/Co3O4)展示了极高的对甲醛的催化氧化活性[13]。但是目前的报道中还缺乏对负载方式影响Ag/Co3O4活性的研究,因此,此研究着重于对原位负载、后浸渍负载以及不负载Ag的介孔催化剂进行对比,考察负载方式对催化活性的影响,并揭示这一影响的内在原因与规律。
1 实验部分
1.1 催化剂的制备
1.1.1模板剂KIT-6的制备
模板剂KIT-6的制备方法已经被广泛报道,将217 g去离子水和11.16 g 质量分数37 %的HCl溶液充分混合并保持温度35 ℃,而后加入6 g P123,搅拌均匀后加入6 g正丁醇搅拌1 h,随之加入12.9 g TEOS持续搅拌24 h。将得到的以上溶液于水热反应釜中在100 ℃条件下反应24 h,将得到的产物用乙醇过滤洗涤3次后于100 ℃下干燥并在550 ℃下焙烧3h,升温速率9 ℃/min。
1.1.2催化剂的制备
原位负载Ag催化剂(Ag/Co3O4-insitu)的制备采用硬模板法,将30 g 无水乙醇,一定含量的Co(NO3)2和AgNO3溶液置于100 mL烧杯中搅拌5 min,而后加入1.288 g KIT-6模板剂并搅拌2 h,将得到的悬浊液置于50 ℃烘箱中干燥24 h,之后将得到的固体置于石英瓶中于450 ℃焙烧3 h。等样品温度降至室温后,按以上方法将其重复浸渍1次。之后得到的样品置于30 mL 2 mol/L的NaOH溶液中在室温下搅拌,刻蚀掉模板剂,重复操作3次后,将样品用水和无水乙醇洗涤各3次,而后置于50 ℃烘箱干燥过夜,之后在450 ℃下焙烧3 h,得到最终产物。不负载Ag的Co3O4催化剂的制备方法与以上基本一致,唯一不同在于在浸渍过程中不加入AgNO3溶液。
后负载Ag催化剂采用浸渍法制备,在得到上述纯Co3O4样品后,将其置于烧杯中加入去离子水,而后加入一定量的AgNO3溶液搅拌1 h。将得到的悬浊液至于旋蒸仪中将水蒸发,得到的产物在干燥过夜后于450 ℃下焙烧3 h,得到最终产物。
1.2 催化剂的表征
X射线衍射谱图(XRD)由型号为X’Pert PRO MPD的X射线衍射仪测量得出。在电压40 kV,电流40 mA条件下,采用Cu_Kα射线(λ=0.15406 nm)在2θ角为10o~80o的范围内以0.02o为步长进行扫描。小角XRD测量条件与广角一致,测量方法为在2θ角为0.5o~5.0o的范围内以0.005o为步长进行扫描。
通过Quantachrome公司的Autosorb-1自动物理吸附仪测定催化剂吸脱附曲线,具体操作流程为:将干燥的催化剂样品在300 ℃下脱气5 h,而后在液氮温度(-196 ℃)下进行N2吸脱附等温线的测定。通过BET方法计算催化剂的比表面积,通过BJH方法计算脱附等温线得出孔容、孔径及其分布。
程序升温还原(H2-TPR)结果由1台装有TCD检测器的化学吸附分析仪(Autochem 2920)测量得到。首先将样品在空气中于300 ℃预处理1 h,之后以50 cm3/min的流速通入体积分数为10% H2/Ar 的混合气;当TCD信号平稳后以10 ℃/min的速率将催化剂本体温度从-50 ℃升温至800 ℃。检测过程中,去除掉升温过程中反应生成的水之后,采用TCD检测器检测相应温度下的氢气消耗量。
X射线光电子能谱(XPS)由AXIS Ultra系统测得,测得的结合能数值经过了C1s(284.8 eV)的校准,该系统采用在225 W和15 kV下产生的Al_Kα射线(hv=1 486.6 eV)激发出光电子。
1.3 催化剂活性评价
催化剂的活性评价在内径为4 mm的固定床石英管中进行,由包裹石英管的管式加热炉升温可以得到所需反应温度。采用一定流量的氮气吹扫多聚甲醛可以产生可控浓度的HCHO/N2混合气体,在评价过程中,我们将甲醛体积分数控制为110×10-6,氧气为20%,氮气为平衡气体。石英管反应器中的催化剂质量为60 mg,混合气流速为100 mL/min,对应空速为100 000 mL/(g·h)。
混合气进、出气体的浓度通过傅里叶变换红外光谱仪(FT-IR,Nicolet iS50)检测,该光谱仪装配有2 m 气体池和硫酸三甘肽晶体(DTGS)检测器,分辨率:0.5 cm-1,光收集区域:4 000~600 cm-1。检测时所取特征振动峰分别为:甲醛2 897 cm-1(C—H振动)和二氧化碳2 350 cm-1(O—C—O振动)。由于没有检测到除二氧化碳外其它的含碳化合物,根据碳守恒我们采用反应后气体中二氧化碳的浓度计算催化转化率。
2 实验结果与讨论
2.1 XRD结果分析
图1a)为3种样品的广角XRD谱图,根据图1中主要的布拉格衍射峰峰位置的查询,可以确定其为Co3O4的各晶面对X射线的衍射(JCPDS 80-1532),这说明样品的主要成分为Co3O4。除Co3O4对应的各个晶面的衍射外,还可以看到单质Ag(JCPDS 87-0597)较弱的XRD峰[图1a)中以黑色菱形标记位置],但没有观察到Ag2O的衍射峰,这使我们确定所制备的样品确为负载型的Ag/Co3O4材料。图1b)为3种样品的小角XRD谱图,可以看出,原位负载的Ag/Co3O4与不负载的Co3O4在2θ角为0.95o左右出现了较为明显的211晶面衍射峰[14],但后负载的样品并未出现该峰,说明其介孔孔道结构经过浸渍后发生了改变。
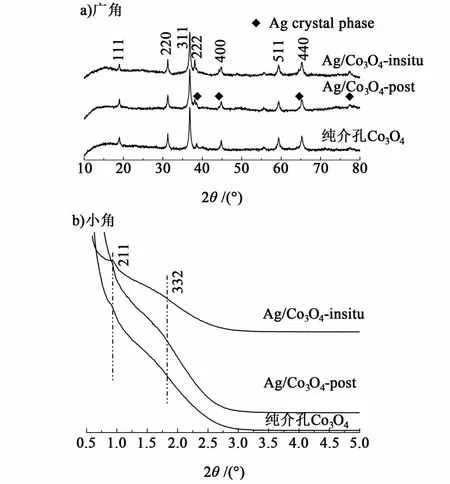
图1 催化剂样品的XRD谱图Fig.1 XRD patterns of the samples
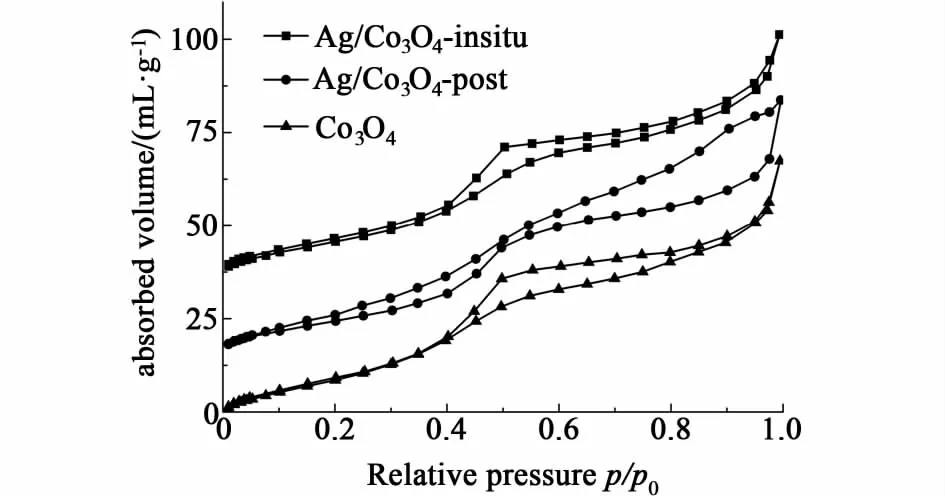
图2 催化剂样品的N2物理吸脱附等温线Fig.2 The isotherms of the samples in N2 physical adsorption & desorption
2.2 氮气物理吸附分析
为了进一步确认根据XRD谱图得出的结论,测取了催化剂样品的氮气等温吸脱附曲线(图2),并计算了样品的表面物理参数。如图2所示,Ag/Co3O4-insitu和纯Co3O4均表现为Ⅳ型等温线,其滞后回线均在相对压力约为0.4处出现,此为介孔材料的明显特征[14]。其中,不负载的纯Co3O4样品的滞后回线相对于Ag/Co3O4-insitu更加完美,说明Ag的负载在一定程度上破坏了介孔结构。与之不同的是后浸渍法负载的样品Ag/Co3O4-post,其吸脱附曲线与前述2种的形态相比发生了巨大的变化,说明浸渍之后,其介孔结构塌陷。表1总结了3种样品的表面物理参数,可以看出3种样品的比表面积、平均孔径、孔容等均相近,但不负载的样品比表面积与孔容均比载Ag的样品相对较高,这进一步说明载Ag后的表面物理性质略微下降,这与吸脱附等温线的结果一致。
2.3 催化剂的活性
证实制备的样品为目标催化剂之后,评价了样品对甲醛的催化活性。图3展示的即为Ag/Co3O4-insitu,Ag/Co3O4-post和纯介孔Co3O4催化氧化甲醛活性随温度的变化曲线,活性评价条件为甲醛体积分数为110×10-6,空速100 000 mL/(g·h)。

表1 样品的比表面积、孔径、孔容与Ag质量分数
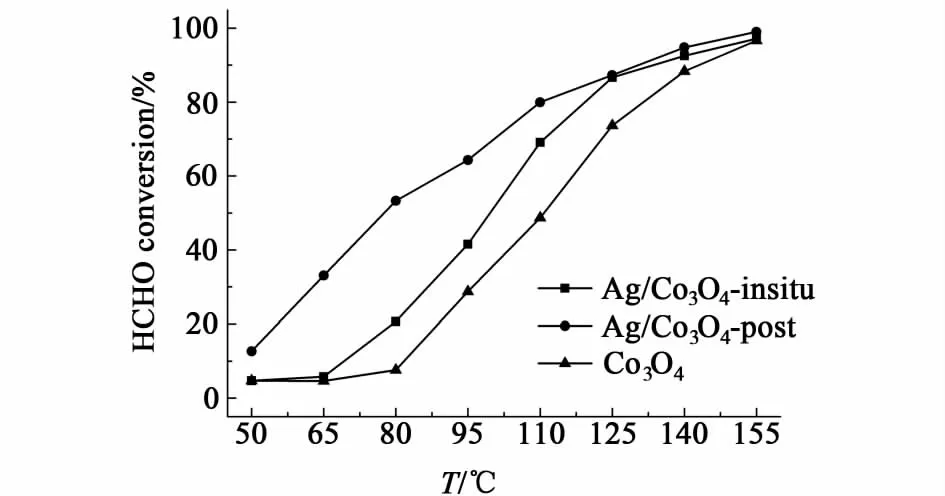
图3 不同负载方法制备的Ag/Co3O4与Co3O4的催化活性图Fig.3 Catalytic activity of Ag/Co3O4with different loading process and the pure mesoporous Co3O4
由图3可以看出,3种催化剂在低温段表现出了巨大的活性差异,其活性顺序由高到低分别为Ag/Co3O4-insitu>Ag/Co3O4-post>Co3O4,这说明一方面Ag的添加有助于活性的提高,另一方面,不同的Ag负载方式也会对活性产生巨大的影响,后负载的方式表现出比原位负载更优良的催化氧化甲醛的活性。通过电感耦合等离子体发射光谱仪(ICP-OES)测取了Ag/Co3O4-insitu和Ag/Co3O4-post的Ag含量,如表1所示,二者Ag质量分数相近,分别为6.01%和7.78%。因此,二者催化活性的巨大差异的主要原因不应是Ag含量的差异,这需要寻找其它影响活性的因素。
2.4 H2-TPR分析
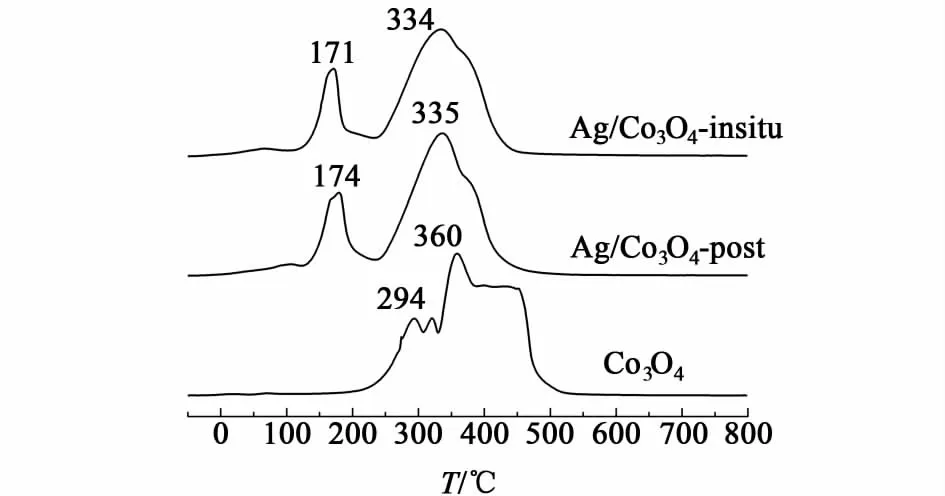
图4 催化剂样品的H2-TPR结果Fig.4 H2-TPR results of the catalysts
为了探究造成催化剂活性差异的原因,首先进行了程序升温还原实验来测试其氧化还原能力,结果见图4。不负载的Co3O4在294和360 ℃出现明显的还原峰,低温还原峰可以归属为Co3+到Co2+还原,高温还原峰为Co2+到Co的还原[15]。类似地,Ag/Co3O4-insitu在171和334 ℃,Ag/Co3O4-post在174和335 ℃出现了还原峰,计算出的对应各峰的氢气消耗量比率为:低温/高温约为1/3,这进一步确认了上述的还原路径。可以看出,Ag的添加使催化剂的还原温度明显降低,进而使负载Ag的催化剂比不负载的具有更高的还原能力,促进了催化活性的提高。
但对于Ag/Co3O4-insitu和Ag/Co3O4-post,其氢气还原步骤与峰型接近一致说明其整体的还原能力相似,催化剂的氧化还原性质并不是影响这2种催化剂催化活性的根本原因,需要从其它角度来探求造成活性差异的原因。由于H2-TPR结果主要反映催化剂的整体化学性质,而催化剂表面所占比率低,因而H2-TPR结果遮盖了表面信息,而表面状态很可能会影响催化反应的速率,因而我们下一步采用X射线光电子能谱来探究表面状态的差异。
2.5 XPS 分析
催化反应主要发生在表面,因而对表面状态的表征更为重要,为了探究催化剂表面的Ag、Co、O物种的状态,进行了X射线光电子能谱分析。图5a)显示的为Ag3d的能谱图,在结合能为374.1和368.1 eV 的峰分别对应表面的Ag3d3/2和Ag3d5/2的谱线[16],3 d谱线双峰的分裂能量为6.0 eV,这些都说明在Ag/Co3O4-insitu和Ag/Co3O4-post样品表面的Ag主要以金属态形式存在;不负载Ag的Co3O4没有Ag3d对应的峰出现,说明表面不存在Ag物种。2种载Ag样品的图谱没有表现出明显的差异,说明不同的负载方式对Ag的电子状态没有明显的影响。
图5b)为Co2p的能谱线和去卷积分峰结果,796.5和781.5 eV的峰分别对应Co2p1/2和Co2p3/2线的Co2+,794.7和779.7 eV的峰分别对应Co2p1/2和Co2p3/2线的Co3+[17-18]。3种催化剂的Co谱线的分峰量化结果和表面Co3+/Co2+物质的量之比列于表2中,结果显示,在3种催化剂表面Co物种呈现出不同的状态,后负载的催化剂表面具有最高的Co3+/Co2+物质的量之比(3.31),原位负载和不负载的催化剂分别为2.95和2.90。这一顺序与活性顺序一致,说明Co3+物质的量之比增加有助于催化氧化甲醛活性的提高。
图5c)展示了3种催化剂表面的O1s谱线与分峰结果图,O1s线可以去卷积为2个代表不同氧物种的峰。在结合能为529.6 eV的主峰可以归为晶格氧(O2-)(标记为Olatt),结合能位于531.1 eV的峰为低配位数的表面吸附氧(O22-或O-)(标记为Oads),如氧缺陷或羟基基团[19-20]。表2中也列出了O的去卷积分峰的结果,可以看出,不同的催化剂表面的O物种状态不同,Ag/Co3O4-post具有最高比率的表面吸附氧(0.68),Ag/Co3O4-insitu和Co3O4的比率则分别为0.64和0.61,这说明表面吸附氧的增多能够促进催化剂的活性。
Xie等[12]认为Co3+为催化氧化CO的活性物种,他们制备的纳米棒Co3O4由于表面暴露出更多的Co3+而极大地提升了其催化活性。Bai等13发现Co3+和氧缺陷的增多也有助于催化剂催化氧化甲醛活性的提高,这与本研究中XPS分析的量化结果一致。Ag/Co3O4-post在浸渍过程中孔道结构塌陷,但孔道的塌陷可能暴露出更多的Co3+和氧缺陷,进而提高了催化活性。
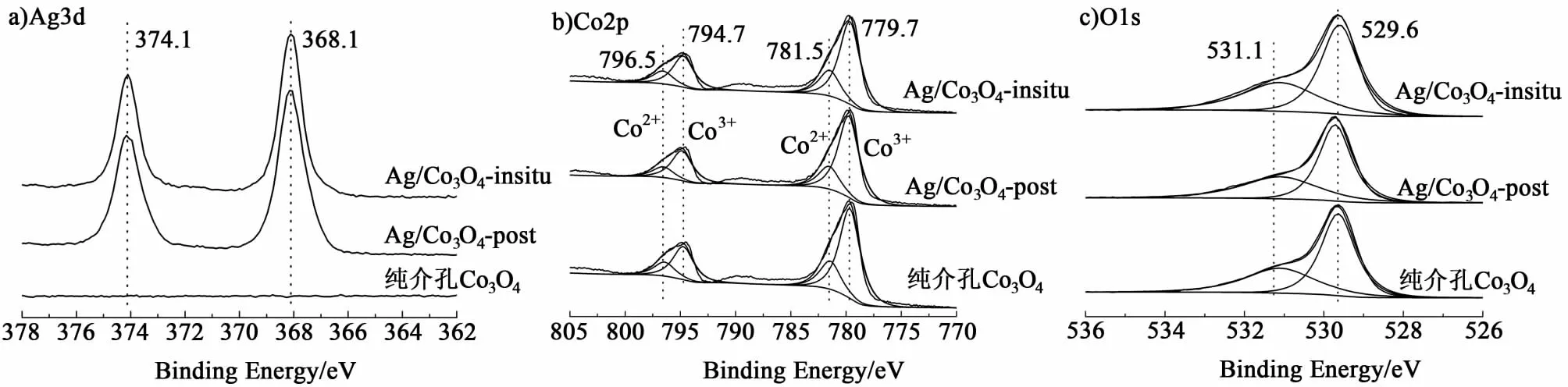
图5 催化剂的XPS谱图Fig.5 XPS spectra of the catalysts
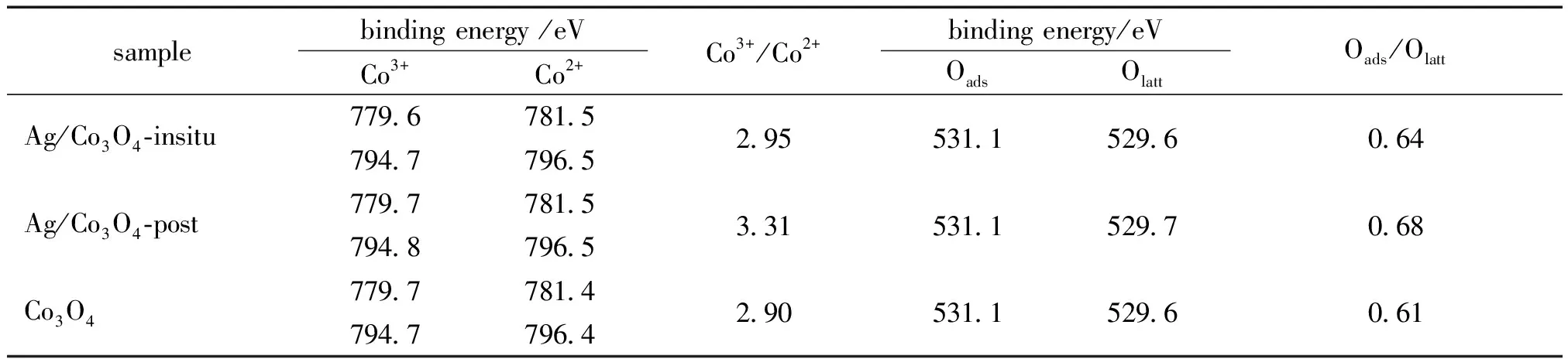
表2 催化剂样品的去卷积分峰结果
3 结论
通过后浸渍法制备的Ag/Co3O4-post由于在制备过程中载体Co3O4的介孔结构发生坍塌,使其暴露出更多的表面Co3+和缺陷氧,同时Ag的担载也提高了样品的还原性能,这均提高了样品催化氧化甲醛的能力,使其表现出在3种催化剂中最优的活性。由于Ag/Co3O4-post在接近室温温度段仍具有一定的催化活性,其改进材料可能具有室温催化氧化甲醛的潜在能力,这对非贵金属催化剂的开发具有一定的指引作用。
参考文献:
[1]Pei J,Zhang J. Critical review of catalytic oxidization and chemisorption methods for indoor formaldehyde removal [J]. HVAC & R Research, 2011,17 (4):476-503
[2]Maddalena R,Russell M,Sullivan D P,etal. Formaldehyde and other volatile organic chemical emissions in four FEMA temporary housing units [J]. Environmental Science & Technology, 2009, 43(15): 5 626-5 632
[3]Yang J,Qin Y,Zeng Y,etal. Progress in endogenous formaldehyde, formaldehyde toxicity and formaldehyde inhibitors [J]. Food Science, 2014, 35 (1):294-297
[4]Osborne H. Non-Iconic abstraction [J]. British Journal of Aesthetics,1976,16 (4): 291-304
[5]Zhang C,Liu F,Zhai Y,etal. Alkali-Metal-Promoted Pt/TiO2opens a more efficient pathway to formaldehyde oxidation at ambient temperatures [J]. Angewandte Chemie International Edition, 2012,51(38):9 628-9 632
[6]Chen B,Shi C,Crocker M,etal. Catalytic removal of formaldehyde at room temperature over supported gold catalysts [J]. Applied Catalysis B: Environmental,2013,(132/133):245-255
[7]Huang H,Leung D Y C. Complete oxidation of formaldehyde at room temperature using TiO2supported metallic Pd nanoparticles [J]. ACS Catalysis,2011,1 (4): 348-354
[8]Zhang C,Li Y,Wang Y,etal. Sodium-Promoted Pd/TiO2for catalytic oxidation of formaldehyde at ambient temperature [J]. Environmental Science & Technology, 2014, 48 (10): 5 816-5 822
[9]Qu Z,Shen S,Chen D,etal. Highly active Ag/SBA-15 catalyst using post-grafting method for formaldehyde oxidation [J]. Journal of Molecular Catalysis A: Chemical, 2012, 356: 171-177
[10]Shi C,Chen B,Li X,etal. Catalytic formaldehyde removal by “storage-oxidation” cycling process over supported silver catalysts [J]. Chemical Engineering Journal,2012,(200/202): 729-737
[11]Zhang C,He H,Tanaka K. Catalytic performance and mechanism of a Pt/TiO2catalyst for the oxidation of formaldehyde at room temperature [J]. Applied Catalysis B: Environmental, 2006, 65 (1/2): 37-43
[12]Xie X,Li Y,Liu Z,etal. Low-Temperature oxidation of CO catalysed by Co3O4nanorods [J]. Nature, 2009, 458 (7 239): 746-749
[13]Bai B,Li J. Positive effects of K+ions on three-dimensional mesoporous Ag/Co3O4catalyst for HCHO oxidation [J]. ACS Catalysis,2014,4 (8):2 753-2 762
[14]Rumplecker A,Kleitz F,Salabas E L,etal. Hard templating pathways for the synthesis of nanostructured porous Co3O4[J]. Chemistry of Materials,2007,19 (3):485-496
[15]Bai B,Arandiyan H,Li J. Comparison of the performance for oxidation of formaldehyde on nano-Co3O4, 2D-Co3O4, and 3D-Co3O4catalysts [J]. Applied Catalysis B: Environmental,2013,(142/143):677-683
[16]Derekaya F B,Güldür Ç. Activity and selectivity of CO oxidation in H2rich stream over the Ag/Co/Ce mixed oxide catalysts [J]. International Journal of Hydrogen Energy,2010,35 (6):2 247-2 261
[17]Tan B J,Klabunde K J,Sherwood P M A. XPS studies of solvated metal atom dispersed catalysts-Evidence for layered cobalt manganese particles on aluninaand silica [J]. Journal of the American Chemical Society,1991,113 (3):855-861
[18]Bonnelle J P,Grimblot J,Dhuysser A. Influence of polarization of bonds on ESCA spectra of cobalt oxides [J]. Journal of Electron Spectroscopy and Related Phenomena,1975,7 (2):151-162
[19]Amri A,Duan X,Yin C Y,etal. Solar absorptance of copper-cobalt oxide thin film coatings with nano-size, grain-like morphology: Optimization and synchrotron radiation XPS studies [J]. Applied Surface Science,2013,275:127-135
[20]Dupin J C,Gonbeau D,Benqlilou-Moudden H,etal. XPS analysis of new lithium cobalt oxide thin-films before and after lithium deintercalation [J]. Thin Solid Films,2001,384 (1):23-32
猜你喜欢
生物学通报(2021年4期)2021-03-16
江苏安全生产(2020年1期)2020-03-16
石油石化绿色低碳(2019年6期)2019-02-13
浙江大学学报(工学版)(2016年11期)2016-06-05
Coco薇(2016年2期)2016-03-22
中国资源综合利用(2016年4期)2016-01-22
河北科技大学学报(2015年5期)2015-03-11
中央民族大学学报(自然科学版)(2014年3期)2014-06-09
中国质量与标准导报(2014年6期)2014-02-28
无机化学学报(2014年4期)2014-02-28
