
异戊烯基二氢黄酮和查尔酮分子结构及光谱的理论计算
2015-03-30 09:34漆文胜孙俊梅李红梅王海峰
成都大学学报(自然科学版) 2015年1期
漆文胜,孙俊梅,李红梅,王海峰
(1.成都大学生物工程学院,四川 成都 610106;2.四川师范大学化学与材料科学学院,四川 成都 610068)
异戊烯基二氢黄酮和查尔酮分子结构及光谱的理论计算
漆文胜1,孙俊梅1,李红梅1,王海峰2
(1.成都大学生物工程学院,四川 成都 610106;2.四川师范大学化学与材料科学学院,四川 成都 610068)
使用密度泛函理论B3LYP方法,对异戊烯基二氢黄酮和查尔酮的分子结构与电子吸收光谱进行理论计算研究,并基于Tomasi的极化统一场模型讨论溶剂效应.结果显示,溶剂对异戊烯基二氢黄酮和查尔酮的前线分子轨道特征影响甚微,溶剂作用使该分子的最大吸收波长红移分别约为5~6 nm和13~15 nm,红移程度与溶剂极性无关.
异戊烯基黄酮;结构;吸收光谱;分子轨道;密度泛函
0 引言
黄酮类化合物广泛存在于各种天然药物和果蔬中,具有较好的抗氧化、抗肿瘤、抗HIV、抗炎、抗血管增生、抗菌和抗疟疾等生物活性,并对人体无毒[1].但从自然界分离出的黄酮远远满足不了人们的需求,近年来,对此类化合物的研究主要集中于天然产物的人工全合成及其生物活性等方面[2-3].二氢黄酮和查尔酮类化合物属于黄酮类化合物,研究发现,合成的二氢黄酮和查尔酮类化合物也具有很好的生物学活性,而其生理活性、作用机制与构效关系可以通过结构修饰来改善,异戊烯基是众多的活性基团中重要的一种[4-5].本研究对异戊烯基二氢黄酮和查尔酮的分子结构与电子吸收光谱进行了理论计算,分析讨论了溶剂效应.此研究做为实验方法的一种有益补充,可得到实验难以获得的结构参数、光谱学等数据,其结果还可加深对异戊烯基黄酮及其衍生物的化学合成、分离提纯等的认识,为进一步从构效关系研讨其药理作用提供理论参考.
1 计算方法
在计算方法上,本研究采用Gaussian 09程序包,由于密度泛函理论综合考虑了电子相关效应,且计算精度高,故首先以此方法在B3LYP/6-311G(d,p)理论水平上对异戊烯基黄酮分子的几种可能结构进行优化,再根据自洽反应场理论的极化统一场模型,利用含时密度泛函理论计算该分子在不同溶剂下的前线分子轨道和电子吸收光谱,最后讨论溶剂效应.
2 结果与讨论
2.1 结构分析
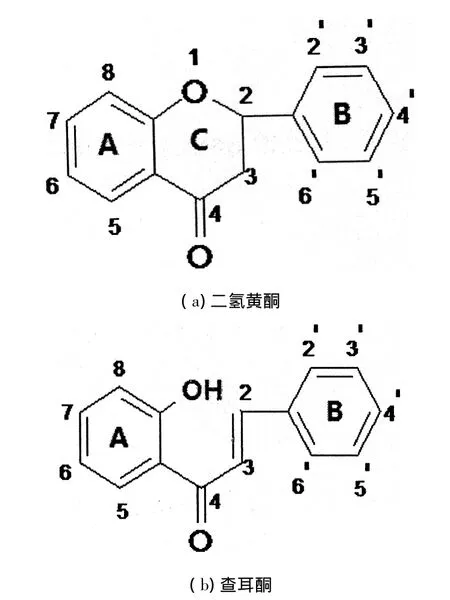
图1 黄酮类化合物结构
黄酮类化合物是由2个苯环(A环与B环)通过3碳链相互连接成的具有6C-3C-6C基本骨架的一系列化合物,其结构特点是分子具有平面多并环共轭的刚性结构,且具有较大的离域键(见图1).有文献报 道 了 对(± )4 '-hydroxy-6,3 ',5 '-triprenyliso-flavonone和(±)-abyssinone-VI 2种天然异戊烯基黄酮的全合成、分离及抗菌性的研究[6-7].在此基础上,本研究分别对2种分子做了进一步的修饰:将前一分子C6上H取代为OH,以下描述为化合物I;将后一分子C6上H取代为异戊烯基,以下描述为化合物II.
图2(a)为化合物Ⅰ的“Y”构象,中间为闭环,其中C2为手性碳,存在R、S构型的手性异构体1R与1S;图2(b)为化合物II的“Y”构象,存在顺反异构体,顺式构型由于羰基与芳环间的强烈位阻效应导致其不如反式稳定,能量高约13 kJ/mol.常温下,通过对化合物Ⅰ和化合物Ⅱ优化后的分子结构进行优化筛选,连在同一个苯环(B环)上的2个异戊烯基空间伸展方向不一样,且能量不同,可将其描述为“Y”和“T”.计算结果表明,“Y”构象比“T”构象能量均低约9 kJ/mol,说明“Y”构象更稳定.另外,互为R、S对映异构体的这2种化合物稳定性相当,这也使得较难对其进行分离提纯操作.因此,本研究主要讨论以异戊烯基“Y”构象为基础的分子并进行计算.

图2 化合物Ⅰ、Ⅱ的“Y”构象
由计算数据可知,化合物Ⅰ的分子整体为非平面型,中间环为半椅式,A环5位酚羟基的键长O2-H约99 pm,比正常值96 pm略微增加,原因是由于空间位阻相对较小,故其键长增加.C5-O2键长为134.1 pm比正常值143 pm略有缩短,原因是该位酚羟基可与羰基O形成分子内氢键O5-H,为169.4 pm.在该氢键的作用下,化合物Ⅰ的A环和氢键形成的环几乎共平面,C5上的羟基与黄酮的主体结构形成了较大的π共轭体系.化合物Ⅱ的A环较结构图发生了翻转,其骨架为经典查尔酮分子骨架,且整个分子结构呈现高度共轭状态,分子中2个苯环(“A”和“B”环)是近乎平面的.O1-H与O5形成了分子内氢键,苯甲酰交叉共轭系统利于母体结构更加稳定,C2=C3与C4=O5桂皮酰交叉共轭系统,使得整个分子共轭体系增大,电子离域更强.此外,化合物Ⅰ和化合物Ⅱ的7位酚羟基O4-H均值为96.3 pm,接近正常值,是其所受的空间位阻较小所致.
2.2 红外光谱分析
物质分子吸收红外线产生的吸收光谱,主要是由于振动和转动能级跃迁引起的,红外吸收光谱不仅可以定性地说明基团的特征吸收频率,而且可以从特征峰的强度来定量讨论,在有机化合物的结构解析上应用很广.本研究在B3LYP/6-311G(d,p)水平下计算了该类化合物的振动频率,计算结果表明,其最小频率均为正值,未出现虚频率,说明所得构象为稳定结构.
化合物Ⅰ、Ⅱ计算结果得到的红外光谱如图3所示.

图3 化合物Ⅰ、Ⅱ的红外光谱
由图3可见,在4 000 cm-1~1 300 cm-1的基团频率区中,3 670 cm-1附近的振动是酚羟基的伸缩振动.3 150 cm-1~3 000 cm-1范围内的振动是异戊烯基上C-H伸缩振动.在1 700 cm-1附近2个分子均出现双键伸缩振动区,强度大、峰尖锐为碳氧双键伸缩振动的特征峰,1 620 cm-1~1 680 cm-1强度稍弱的为C=C的伸缩振动区,芳环上C=C伸缩振动在1 600 cm-1左右.在1 692 cm-1出现了C5上的羟基与黄烷酮的主体结构形成了较大的π共轭体系的骨架振动,此进一步说明形成了较强的分子内氢键.此外,1 300 cm-1~600 cm-1的指纹区内,1 260 cm-1~1 000 cm-1为分子中的碳氧(C-O)伸缩振动吸收峰.900 cm-1~600 cm-1处为 Ar-H 的面外弯曲振动,也存在化合物Ⅰ中C-O单键的伸缩振动.
2.3 紫外光谱与前线分子轨道分析
黄酮多呈黄色,且颜色与分子中的交叉共轭体系(发色团)及助色团有关,在紫外灯光下普遍具有荧光.
通过计算化合物Ⅰ及Ⅱ在气相及乙酸乙酯、甲醇、水溶剂下的50个单重—单重激发态、HOMO、LUMO轨道本征能量和电子吸收光谱中的最大吸收波长,以此来讨论溶剂效应(见表1、2).

表1 异戊烯基黄酮在气相和不同溶剂下的电子吸收光谱数据(化合物I)

表2 异戊烯基黄酮在气相和不同溶剂下的电子吸收光谱数据(化合物II)
化合物Ⅰ和化合物Ⅱ的紫外光谱由2个主要吸收带组成:300~400 nm区间,由B环桂皮酰系统的电子跃迁所引起;240~285 nm区间,由A环苯甲酰系统的电子跃迁所引起.化合物Ⅰ和化合物Ⅱ在气相中的最大吸收波长(最低能量吸收波长)分别为325.7 nm和362.3 nm,属于近紫外区,电子跃迁形式均为电子的HOMO轨道向LUMO轨道的π-π*跃迁.化合物Ⅱ的S0-S1的π-π*跃迁吸收峰比化合物Ⅰ的强很多,这应该是由于化合物Ⅱ中C2=C3为双键,共轭双键数目越多,与黄烷酮的主体结构形成了更大的π共轭体系,其助色基团的效应也越强,导致共轭体系的π→π*跃迁所产生的吸收带K带向长波方向移动,其值为13~15 nm,比化合物Ⅰ的红移值5~6 nm要大.此外,不同极性溶剂的使用对化合物Ⅰ和II的最低能量吸收波长影响甚微.
此外,在UV光谱中,化合物Ⅰ在270 nm附近有一个较强的、主要由HOMO-4向LUMO的电子跃迁导致的吸收峰,溶剂的使用使得该吸收峰红移了5 nm左右,但溶剂的极性对该吸收峰无明显影响.化合物Ⅱ在280附近也有一个不明显的吸收峰,因为在C=O上O带有未成键电子,n→π*跃迁为禁阻跃迁,出现弱吸收带,溶剂使用出现紫移2 nm左右.在210~220 nm范围内的最强吸收峰为电子的多个能级跃迁吸收峰的叠加,可能是生色团、助色团之间的空间过于拥挤,导致共轭程度降低,吸收峰紫移1~5 nm左右,但均值较小.溶剂极性越强,紫移值则略有增大.
通常,有机分子的电子光谱与前线区域轨道能量,尤其是HOMO与LUMO之间的能级差△E有很大关系.黄酮类化合物具有较大的共轭键,其几何构型与电子光谱之间存在着密切的关系,研究化合物的电子光谱有利于推测和确定其空间构型.
由表1、2数据可见,这2个异戊烯基黄酮分子无论在气相还是在液相溶剂中,HOMO电子云主要集中在成键π轨道上,LUMO电子云则主要分布于共轭反键π*轨道上,化合物II的HOMO轨道上的电子云分散得比较均匀,使得异戊烯基与黄烷酮的主体形成了较大的π共轭体系,且溶剂对化合物Ⅰ及化合物II的HOMO、LUMO轨道上的电子云分布均无明显影响.
3 结论
本研究采用密度泛函理论B3LYP方法对异戊烯基黄酮分子的结构、前线分子轨道以及电子吸收光谱进行了理论计算研究,并得到如下结论:酚羟基与羰基O形成分子内氢键使得分子的稳定性增加,C=C与主体结构形成了更大的共轭体系利于发色与助色效应,溶剂对异戊烯基黄酮的前线分子轨道特征几乎无影响,溶剂效应使2种分子的最大吸收波长红移约5~6 nm和13~15 nm,即π→π*跃迁红移,n→π*跃迁紫移,但程度与溶剂的极性大小影响甚微.
[1]陈永钧,龙晓英,潘素静,等.黄酮类化合物的药效机制及构效关系研究进展[J].中国实验方剂学杂志,2013,19(11):337-344.
[2]杨金会,谢一民,冯尚彪,等.四种天然双异戊烯基黄酮的合成及其抑菌活性研究[J].有机化学,2013,33(10):2155-2161.
[3]赵平,张颖君,山本浩文,等.苦参异戊烯基黄酮类化合物的化学,活性及其生物合成研究进展[J].天然产物研究与开发,2004,16(2):172 -178.
[4]张茵,查晓明,尚靖,等.天然二氢黄酮类化合物研究进展[J].亚太传统医药,2013,9(8):65 -69.
[5]常军,王晨曦,李玉萍.天然黄酮类化合物的构效关系最新研究进展[J].天然产物研究与开发,2013,25(7):1006-1010.
[6]聂汉,李权,赵可清.西红花酸二甲酯电子吸收光谱与热力学性质的理论研究[J].有机化学,2012,32(1):121-126.
[7]吴建章,李物兰,陈玲姿,等.查尔酮及其螺杂环衍生物的合成、晶体结构、抗氧化活性研究[J].有机化学,2012,32(11):2141 -2147.
Theoretic Calculation on Structures and Spectra of Prenylated Dihydroflavonone and Chalcone
QI Wensheng1,SUN Junmei1,LI Hongmei1,WANG Haifeng2
(1.School of Bioengineering,Chengdu University,Chengdu 610106,China;2.College of Chemistry and Materials Science,Sichuan Normal University,Chengdu 610068,China)
The structures and electronic absorption spectra of Prenylated Flavonoids and Chalcone are calculated by the Density Functional Theory(DFT)B3LYP method with 6-311G(d,p)basis set.The polarized continuum models of tomasi(PCM)have been used to discuss the solvent effects.The computational results show that the solvents have little influence on the frontier molecular orbital features of Prenylated Flavonoids and Chalcone.The solvents action makes the maximum absorption wavelength red-shift about 5~6 nm and 13~15 nm respectively.The red-shift is independent of the solvent polarity.
Prenylated Flavonoids;structure;absorption spectrum;frontier molecular orbital;density functional theory
O641;O622.4
A
1004-5422(2015)01-0001-04
2014-12-16.
漆文胜(1968—),男,硕士,副教授,从事物理化学理论计算研究.
猜你喜欢
世界科学技术-中医药现代化(2021年10期)2021-03-02
天然产物研究与开发(2018年10期)2018-11-06
天然产物研究与开发(2018年10期)2018-11-06
中成药(2018年9期)2018-10-09
中国资源综合利用(2017年1期)2018-01-22
山东工业技术(2016年15期)2016-12-01
中国粮油学报(2016年5期)2016-01-23
中国医学科学院学报(2015年5期)2015-03-01
天然产物研究与开发(2014年6期)2014-04-27
郑州大学学报(理学版)(2014年3期)2014-03-01
