
DNA作催化剂:小分子体外不对称催化合成
2015-03-21 11:29冯凯波
生物学杂志 2015年1期
卢 彦, 冯凯波
(1. 南京大学 生命科院学院, 南京 210093; 2. 南京大学 化学化工学院化学系, 南京 210093)
DNA作催化剂:小分子体外不对称催化合成
卢 彦1, 冯凯波2
(1. 南京大学 生命科院学院, 南京 210093; 2. 南京大学 化学化工学院化学系, 南京 210093)
综述了以插入非手性二价铜配合物的鲑鱼精DNA(st-DNA)作为催化剂,于体外催化包括不对称Diels-Alder反应、Friedel-Crafts反应与Michael加成等一系列小分子反应的研究进展。这些反应在天然产物与其他具生物活性物质合成途径设计中有重要作用。反应通常可以取得大于80%的对映体过量百分数(ee)值,并于优化后可进一步提高到90%至99%。DNA的碱基序列对对映选择性将产生影响,且因配体结构而异,有较多G或C相连的DNA序列通常可以产生较好的对映选择性。
DNA;不对称;催化;插入;加成
天然产物等一干具有生物活性的分子皆具有多个手性中心,通过传统合成方式将得到外消旋化合物,并无法满足生物医药上的用途,欧洲曾发生的外消旋反应停致畸事件即为例证。若是对其进行不对称拆分,则理论上每次至少损失50%的产物,产率低下而于原子经济性有极为不利的影响。作为解决之道的不对称催化近年来成为有机化学中的前沿研究领域。
催化剂的选取对反应成败至关重要。通常用于不对称催化的催化剂为如Mn-Salen的过渡金属的手性配合物,除少数外通常具有复杂结构而较难获取或价格昂贵,而对合成路线设计造成负面影响[1-3]。也有采取不含金属中心的手性配体,包括如天然氨基酸L-脯氨酸及其衍生物。这一类催化剂通常结构更加简单,但也需经过一些修饰以达到高的选择性,如L-脯氨酸上羧基常被修饰为三甲硅氧基二苯基-L-脯氨醇[4-7],具体如图1所示。

图1 基于L-脯氨酸之手性催化剂
自然界常进行高效高选择性的不对称合成,如天然产物紫杉醇的合成,常常超出化学手段的效率。其过程的复杂性使得现阶段尚未能够实现对自然界合成方法的体外重复,但为模仿自然手段进行不对称催化,可以考虑选用自然界中含量丰富的手性物质,于常温常压、水作为溶剂下进行手性诱导,以期取得较人工合成催化剂选择性更高的催化反应。DNA作为生物体内遗传信息的载体大量存在,且有天然具备之右手双螺旋结构,正好可以满足模仿天然过程的需要。
2005年Roelfes与Feringa首度报道以插入平面二价铜非手性配合物的DNA为催化剂进行的不对称Diels-Alder反应[8],此后这一催化过程即被广泛研究。本文将回顾自此以来对DNA催化各类小分子反应的研究进展,并对其未来发展方向作一展望。
1 催化机制
DNA虽然具有天然的右手双螺旋结构,但分子内的核苷上2位因脱氧而造成可能与底物的结合位点减少,也使得其难以如RNA形成具有催化活性的三级结构[9],具体如图2。

图2 RNA与DNA氢键作用力比较
因此,需要在DNA分子中引入合适的结合位点,方能使其与底物作用而通过其二级结构所具有的手性特征进行不对称催化。Roelfes与合作者将含有氮原子的平面芳香型小分子与鲑鱼精DNA(st-DNA)作用,这些分子可以插入DNA的碱基序列间,从而提供两个氮原子作为结合位点[8, 10],具体如图3所示。
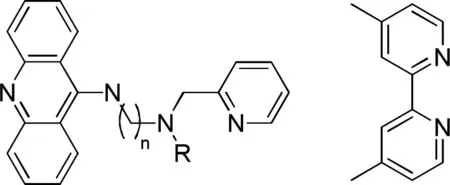
图3 第1代(左)与第2代(右)非手性配体
这些配体并不需具有手性,因此使用它们更具经济性。底物为具有类似图4结构的α,β-不饱和酮,也具有氮氧原子可以连接性;但配体的氮原子无法与底物直接相连,因此引入铜作为中心原子,通过四配位的方式连接底物以进行催化[8],具体如图4所示。
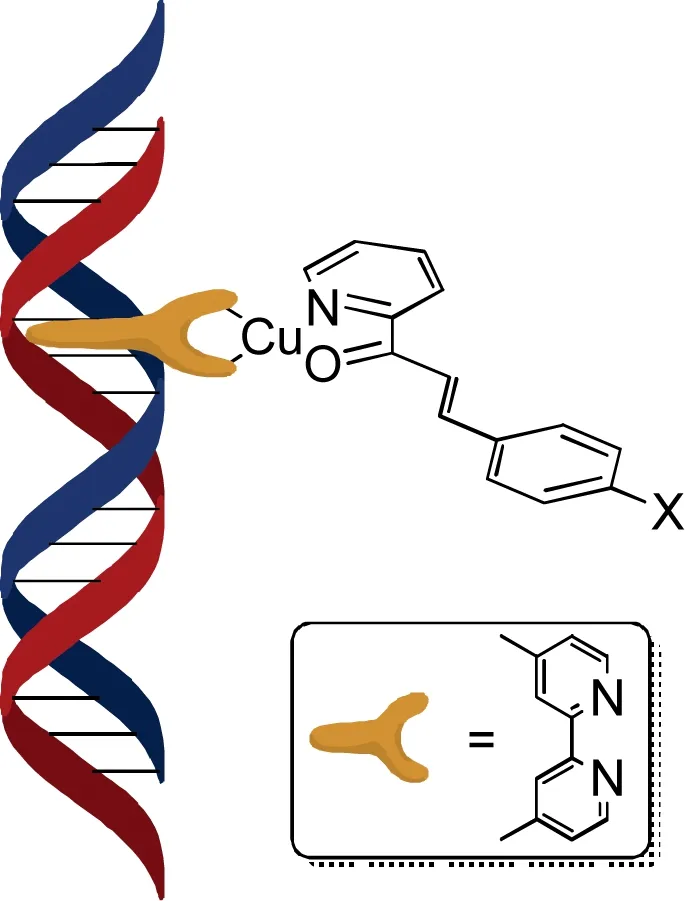
图4 插入配合物之DNA
此时底物分子即被拉到DNA分子内部而受其手性诱导。DNA的双螺旋结构将在底物平面的Re面产生一位阻,而使得亲核试剂等只可从另一面的Si面进攻,从而实现不对称催化[11],具体如图5所示。

图5 DNA诱导手性催化机制
由于底物已经与DNA结合,任何可以与底物反应的试剂均可比照对底物进攻,从而得到各种不同种类的手性反应产物,为合成路线设计带来很大便利。
2 不对称催化
2.1 Diels-Alder反应
不对称Diels-Alder反应是最先被报道的催化反应[8]。底物亲双烯体如前所述,为α,β-不饱和酮,分子中双键与双烯体环戊二烯作用而得到手性产物[8, 10]。其中R基处在分子外部,对反应并无太大影响,可为烷基、苯基等多种基团;当使用第1代催化剂时,主要的内型(endo-)产物对映体过量百分数(ee)最高仅可达到53%[8], 但改用第2代配体,ee值即提升至超过99%[10], 使得此一反应始具有实际的应用价值。此一提升系因用于与铜离子配位的氮在第2代配体中被安置在插入DNA碱基序列的芳环中,从而使铜离子以及与其配位的底物更接近DNA的双螺旋结构(见图6),于反应时产生更大位阻,而致更佳的立体选择性[10]。
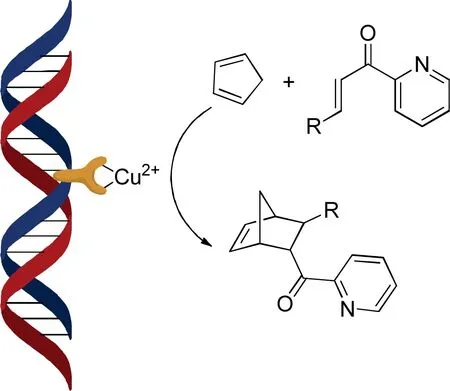
图6 DNA催化不对称Diels-Alder反应
底物羰基另一侧所接基团并不限于吡啶,在改接咪唑等其他含氮杂环后,也可以达到近似同等的催化效率,其ee值在98%上下,也具实用价值[12],可见其对底物并不具有苛刻的选择性,催化反应可以设计的空间较大。
Chandra与Silverman在2008年报道了以DNA的5′末端经醚键连接蒽环而诱导的一类Diels-Alder反应[13],与Roelfes的催化机制不同。但由于系以DNA尾端经氧杂长链连接底物,未能有效利用DNA的空间结构,因此也未能产生ee值,此处便不详述。
2.2 Friedel-Crafts反应
对于Friedel-Crafts反应的研究相对较少,其过程与前并无二致,惟双烯体改为由杂环芳烃充当,发生反应如图7所示[14]。
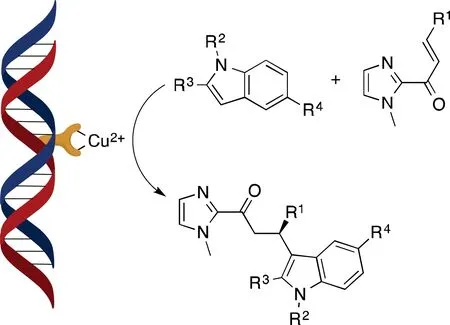
图7 DNA催化不对称Friedel-Crafts反应
反应仍然沿用第2代配体,其ee值最高可达到93%[14]。4个取代位点可接各类烃基,从而使反应范围进一步扩大。
前已提及反应需在水相中进行,但由反应物结构可知其在水相中溶解度欠佳,从而使反应速率受到影响。为解决此问题,Rosati与合作者在反应体系中添加十二磺基硫酸钠(SDS)胶束为乳化剂,使反应速率大幅提升,速率常数最高可提升1.2×103倍,而使反应更具有实用价值[15]。关于Friedel-Crafts反应的更多细节,包括反应条件的进一步优化与底物拓展还有待未来研究。
2.3 Michael加成
底物所具有的α,β-不饱和羰基化合物结构使其能够被各类亲核试剂进攻而在其β位接入一个取代基。这一反应可以产生各类手性产物,较前两类反应应用价值更大。反应通式如图8所示[11]。

图8 DNA催化不对称Michael加成
Roelfes等人试验了一系列的亲核试剂,包括硝基甲烷与丙二酸二乙酯[11]、水[16]、醇[17]等,多数可以取得良好对映选择性而得到高的ee值,最高可至99%,而以水与醇为亲核试剂的反应其选择性略差[11],但也可达到82%与86%左右。与Friedel-Crafts反应不同的是,DNA的存在虽然使得不对称合成成为可能,但却并不一定加快反应速率,反而有时可能降低速率,如使用硝基甲烷为亲核试剂时[18]。
以水为亲核试剂的反应提供了β-羟基酮的建构方式,且对底物结构并无苛刻要求;这一模体在许多天然产物如紫杉醇中均有存在,且以水为溶剂,非蛋白类酶催化也是前所未见。这些反应还有进一步优化的空间,而亲核试剂的范围也可以继续扩大,未来可预期还将有更多进展。
2.4 反应条件优化
最初应用在催化反应上的DNA为未经筛选的st-DNA,其序列对对应选择性所可能产生的影响并不明确[8, 10];2008年Boersma及合作者首度研究了碱基序列对选择性的影响。当选用第2代配体,寡核苷酸回文序列时[19],对于图6所示的不对称Diels-Alder反应有如下关系[19],见表1。
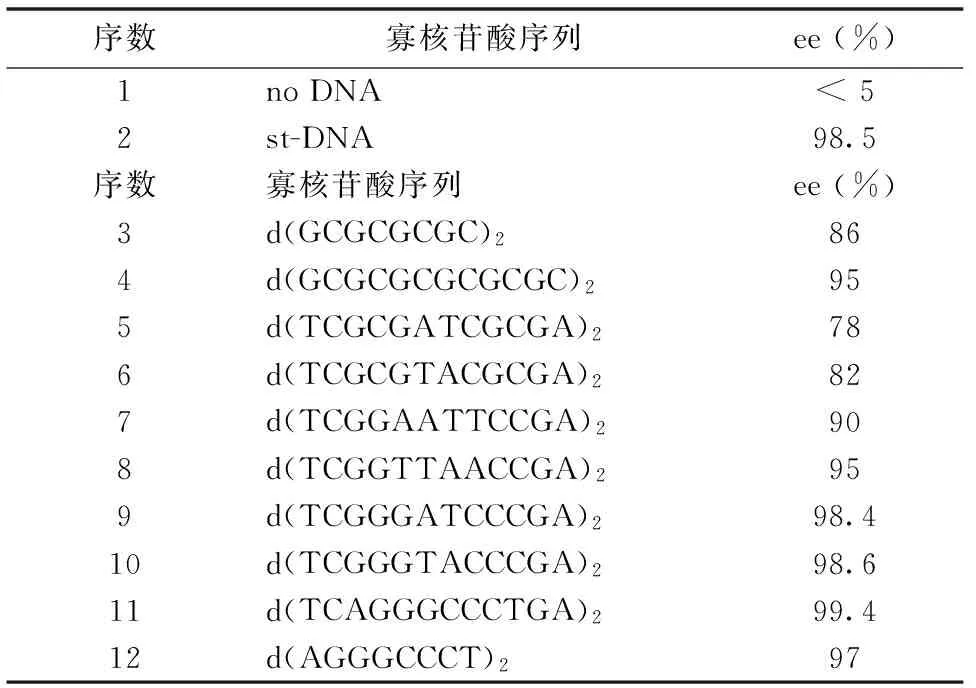
表1 st-DNA序列与ee值关系(部分)
可见所获取的ee值与碱基序列中G、C的含量呈正相关。当序列中的G或C成单出现时,ee值仅有80%上下,而出现二连G与C时,ee值即升上90%~95%,而至三连G与C时则升至98%以上,再移除序列中的AT中心后即最终提至99%以上。单链DNA也可进行不对称催化,但效果较差而不宜采用。文献未报道连续存在的C或G造成高选择性的原因[19],但因GC之间氢键较AT多出1对,可能与插入的配体相互作用更强而使其更加紧密结合;同时连续的C或G也使DNA结构转变而综合作用所致。这与第1代配体的选择偏好稍有不同,但在之后的报道中可看出高含量的G与C亦对选择性有所助益[19-20]。
即使DNA的右旋结构已经确定,通过对配体结构的改变仍然可以控制所得手性产物的R/S构型。对不对称Diels-Alder反应,采取下面的配合物可以得到与采用第2代配体相反的手性构型[21]。

图9 产生异常选择性的配合物
这一配体中的Cu采取八面体配位,因而构型与前不同,可能因此造成其与底物结合后使其取向发生改变而得到相反的构型[21]。其最高可达到-92%的ee值[21], 有一定的应用价值。
之前的反应中试剂用量均停留在小剂量如微克级别[8], 无法满足正常合成的需要。2009年Megans与Roelfes将反应扩展至克级,可在水与有机溶剂如乙腈中进行而得到最高96%的ee值,并可应用于Diels-Alder、Friedel-Crafts反应与Michael加成,使这一反应真正具有了实用上的意义,并可能作为未来进行药物合成的手段[22]。
3 展望
以铜配合物插入DNA而进行的一系列不对称催化现在已经发展得较为成熟,未来工作应着重于底物的拓展以及更多可用于反应试剂的研究。此催化的局限主要在于底物结构中须有可与铜配位的氮氧结构。若有不具此类结构的底物,则仍需采取传统不对称催化手段,或设法引入此类结构,如在羰基之α碳上引入含氮基团。且现阶段Michael加成反应所试验的亲核试剂尚且种类较少,有待于未来的进一步拓宽,尤其若是将NH3或其衍生物作为亲核试剂则可能得到β-氨基酮,有机会将其脱杂环并氧化为β-氨基酸,这类物质多具有生物活性,将在包括药物合成与设计的领域中前景光明。现阶段既已明了对催化有利的DNA碱基序列,可利用PCR技术大量合成具有相应序列的DNA,既然反应已经扩展到克级,若稍加优化可能即可确实进行生产,而免去采取各类复杂手性催化剂的麻烦,其应用值得期待。
[1]Jacobsen E N, Zhang W, Muci A R, et al. Highly enantioselective epoxidation catalysts derived from 1,2-diaminocyclohexane [J]. Journal of the American Chemical Society, 1991, 113(18): 7063-7064.
[2]Cai Y, Liu X, Hui Y, et al. Catalytic asymmetric bromoamination of chalcones: highly efficient synthesis of chiral α-bromo-β-amino ketone derivatives [J]. Angewandte Chemie, 2010, 122(35): 6296-6300.
[3]Cai Y, Liu X, Jiang J, et al. Catalytic asymmetric chloroamination reaction of alpha,beta-unsaturated gamma-keto esters and chalcones [J]. J Am Chem Soc, 2011, 133(15): 5636-5639.
[4]Appayee C, Brenner-Moyer S E. Organocatalytic enantioselective olefin aminofluorination [J]. Org Lett, 2010, 12(15): 3356-3359.
[5]Deiana L, Dziedzic P, Zhao G L, et al. Catalytic asymmetric aziridination of α,β-unsaturated aldehydes [J]. Chemistry-A European Journal, 2011, 17(28): 7904-7917.
[6]Rios R, Sund N H, Ibrahem I, et al. Highly enantioselective synthesis of 2H-1-benzothiopyrans by a catalytic domino reaction [J]. Tetrahedron Letters, 2006, 47(48): 8547-8551.
[7]Vesely J, Ibrahem I, Zhao G L, et al. Organocatalytic enantioselective aziridination of α,β-unsaturated aldehydes [J]. Angewandte Chemie, 2007, 119(5): 792-795.
[8]Roelfes G, Feringa B L. DNA-based asymmetric catalysis [J]. Angewandte Chemie International Edition, 2005, 44(21): 3230-3232.
[9]Silverman S K. Deoxyribozymes: DNA catalysts for bioorganic chemistry [J]. Organic & Biomolecular Chemistry, 2004, 2(19): 2701-2706.
[10]Roelfes G, Boersma A J, Ferringa B L. Highly enantioselective DNA-based catalysis [J]. Chemical Communications, 2006(6): 635-637.
[11]Coquire D, Feringa B L, Roelfes G. DNA-based catalytic enantioselective Michael reactions in water [J]. Angewandte Chemie International Edition, 2007, 46(48): 9308-9311.
[12]Boersma A J, Feringa B L, Roelfes G. α,β-unsaturated 2-acyl imidazoles as a practical class of dienophiles for the DNA-based catalytic asymmetric Diels?Alder reaction in water [J]. Organic Letters, 2007, 9(18): 3647-3650.
[13]Chandra M, Silverman S K. DNA and RNA can be equally efficient catalysts for carbon carbon bond formation [J]. Journal of the American Chemical Society, 2008, 130(10): 2936-2937.
[14]Boersma A J, Feringa B L, Roelfes G. Enantioselective Friedel-crafts reactions in water using a DNA-based catalyst [J]. Angewandte Chemie International Edition, 2009, 48(18): 3346-3348.
[15]Rosati F, Oelerich J, Roelfes G. Dramatic micellar rate enhancement of the Cu2+catalyzed vinologous Friedel-crafts alkylation in water [J]. Chemical Communications, 2010, 46(41): 7804-7806.
[16]Boersma A J, Coquire D, Geedink D, et al. Catalytic enantioselective syn hydration of enones in water using a DNA-based catalyst [J]. Nature Chemistry, 2010, 2(11): 991-995.
[17]Megensa R P, Roelfes G. DNA-based catalytic enantioselective intermolecular oxa-Michael addition reactions [J]. Chemical Communications, 2012, 48(51): 6366-6368.
[18]Dijk E W, Boersma A J, Feringa B L, et al. On the role of DNA in DNA-based catalytic enantioselective conjugate addition reactions [J]. Organic & Biomolecular Chemistry, 2010, 8(17): 3868.
[19]Boersma A J, Klijn J E, Feringa B L, et al. DNA-based asymmetric catalysis: sequence-dependent rate acceleration and enantioselectivity [J]. Journal of the American Chemical Society, 2008, 130(35): 11783-11790.
[20]Rosati F, Boersma A J, Klijn J E, et al. A Kinetic and structural investigation of DNA-based asymmetric catalysis using first-generation ligands [J]. Chemistry-A European Journal, 2009, 15(37): 9596-9605.
[21]Boersma A J, De Bruin B, Feringa B L, et al. Ligand denticity controls enantiomeric preference in DNA-based asymmetric catalysis [J]. Chemical Communications, 2012, 48(18): 2394-2396.
[22]Megens R P, Roelfes G. Organic co-solvents in aqueous DNA-based asymmetric catalysis [J]. Organic & Biomolecular Chemistry, 2010, 8(6): 1387-1393.
DNA as catalyst: catalyticinvitroasymmetric syntheses of small molecules
LU Yan1, FENG Kai-bo2
(1. School of Life Science, Nanjing University, Nanjing 210093, China; 2. Department of Chemistry,School of Chemistry and Chemical Engineering, Nanjing university, Nanjing 210093; )
This paper reviews recent works oninvitroasymmetric catalysis of small molecules, with salmon testes DNA (st-DNA) inserted by achiral Cu(II) complexes as catalysts. Possible reaction types include Diels-Alder Reaction, Friedel-Crafts Reaction and Michael Addition that are crucial for synthetic route design of natural products and other biologically active compounds. These reactions usually obtain fairly high enantiomeric excess (ee) of over 80%. After optimization, the ee value can further increase to 90% to 99%. The base sequence of DNA will affect the enantioselectivity in ways differ with the ligand structure. Generally, a DNA molecule with more continuous G or C sequence has better enantioselectivity.
DNA; asymmetric; catalysis; insertion; addition
2014-06-25;
2014-08-01
国家基础科学人才培养基金(NSFC J1103512)
冯凯波,专业方向为有机合成方法学,E-mail: 517down@gmail.com;
卢 彦,博士,讲师,主要从事分子生物学研究,E-mail:luyan@nju.edu.cn。
O621.3+5;O643.36
A
2095-1736(2015)01-0086-04
doi∶10.3969/j.issn.2095-1736.2015.01.086
猜你喜欢
分子催化(2022年1期)2022-11-02
科学(2020年2期)2020-08-24
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
海南师范大学学报(自然科学版)(2015年2期)2015-12-23
山西建筑(2014年6期)2014-11-09
重庆三峡学院学报(2014年3期)2014-07-16
郑州大学学报(理学版)(2014年3期)2014-03-01
大学化学(2014年1期)2014-01-26
天津医科大学学报(2011年4期)2011-07-13
