
Ga12-nAsn(n=0~12)系列团簇基态结构及稳定性的研究
2015-03-20 01:13:28潘仁奇朱挥毫王秀敏郭颖旦杨建宋
杭州师范大学学报(自然科学版) 2015年4期
潘仁奇,姜 科,朱挥毫,王秀敏,郭颖旦,杨建宋
(1.杭州师范大学经亨颐学院,浙江 杭州311121;2.杭州师范大学理学院,浙江 杭州310036)
0 引言
早在20世纪80年代,开始从理论和实验两个层面,对纳米尺度的砷化镓团簇物理性质进行研究[1-2].理论的研究基于第一性原理,常借助一些量子计算平台.1993年,Mohammad 等用ab-initio方法研究了Ga4As4团簇[3-4],1991年,Lou等用Dmol方法研究了Ga5As5团簇[5],1999年,Yi用Car-Parrinello方法研究了Ga6As6团簇,认为其基态结构是一个带双帽的立方结构[6].2000年后,赵继军等用Dmol软件、Karamanis等用B3LYP/cc-p VTZ-PP方法对GanAsn(2≤n≤9)团簇进行计算后发表了多个相应的基态结构[7-8],也揭示了原子极化率等物理性质随团簇大小的演变规律.2000年后,赵伟等用全势能线性Muffin-tin轨道分子动力学方法(FP-LMTO-MD)也对GanAsn(n=4~6,8)做过计算和分析,提出过一些能量更低的结构[9-12].同样,对带电的砷化镓离子团簇,1986年,Smally等用激光蒸发技术产生Ga As中性团簇和它们的正负离子团簇[13].2001年,Taylor等对和负离子团簇进行过光电子谱的研究[14].2005年,Zhu用CASSCF/DFT/CCSD(T)方法研究过Ga2As2、团簇的低电子态光谱性质[15].2008年,Gutsev等用广义梯度近似下的密度泛函理论对中性的、带正、负电的GanAsn(n=2~15)团簇结构和能量展开过计算[16-17].2005—2009年间,我们组也曾经用FP-LMTO-MD 方法仔细研究过GanAsn(n=4~7)离子团簇[18-19].
对砷和镓原子数目不对等的GamAsn团簇,近年来也引起了人们研究的兴趣.早在1995年,Liao等发表过关于Ga3As2和Ga2As3团簇电子结构的文章[20].2008年前后,马德明利用密度泛函理论(DFT)对GamAsn(m=1、2;n=1~5)团簇结构和稳定性进行过系统的研究[21-22],得到了GamAsn(m=1、2;n=1~5)的基态和亚稳态结构,并发现团簇的稳定性随原子数的增多而增强;在总原子数目一样的团簇中,砷原子多的团簇将比镓原子多的团簇结合能更大一些;同时也发现团簇的HOMO-LUMO 能隙随原子数的增加呈奇偶交替的变化.我们组在2013年也用ADF软件计算过总数为9个原子的砷化镓系列团簇Ga9-nAsn(n=0~9),发现在n=5和6时,团簇的稳定性比较好[23].类似的研究在硅化铝、氮化镓和磷化镓等团簇中也有所开展[24].
本文将采用基于第一性原理,在阿姆斯特丹密度泛函程序(ADF)平台上,对Ga12-nAsn(n=0~12)系列团簇的基态结构进行随机筛选,选出低能量的结构对其电离能等其他物理性质展开精确计算.目标是找出其基态结构,并弄清楚能量、能隙等物理性质随团簇成分变化而发生的变化规律.
1 计算方法和平台
计算方法分两个阶段,首先利用丁望峰博士改编的随机计算程序auto1.2对Ga12-nAsn(n=0~12)团簇进行较大范围的随机筛选,程序对团簇的初始空间构形设计了3种较典型的情况:即球状的、笼状的和盒状的.球状的和盒状的构形设计首先要考虑两个因素,即避免重叠和避免发生散包现象.由程序自动地在这样一个球(或盒)内建立起Ga12-nAsn(n=0~12)团簇初始配置的原子位置.而笼状结构与球状结构不一样的就是把团簇中的所有原子都安排在一个球的表面.利用随机计算我们获得了数以万计的团簇结构,每一个结构都由计算机进行数万次的迭代优化完成.通过对这些结构的筛选,找出能量比较低的结构.
精确计算的结构的初始构形有两个来源:一是上述初筛出的低能量的结构;二是利用对称性或利用前人已经求得的一些低能态结构通过替代或吸附的方法来构造Ga12-nAsn(n=0~12)的初始几何构形.替代指的是用别种原子去替代某一稳定结构中的某一个原子;吸附指的是在已有的Ga11-nAsn基态结构上,增加一个As原子或Ga原子使之成为新的Ga12-nAsn团簇,再去探求能量最低的Ga12-nAsn(n=0~12)的结构.
有了这些Ga12-nAsn(n=0~12)团簇的初始几何构形,在ADF 密度泛函程序平台上进行几何优化.通过频率计算剔除了所有不稳定的结构后,所得的Ga12-nAsn(n=0~12)团簇的最低能量结构被视作了该类团簇的基态结构.在此基础上,再进一步分析Ga12-nAsn(n=0~12)团簇基态时的其他物理性质,比如绝热电离势(IPs)、绝热电子亲和势(EAs),以及最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)之间的能隙(Eg);分析团簇基态的总能量和关联能随着这两种原子成分比的一般变化规律,同时也计算了团簇能量的一阶和两阶差分,分析团簇的稳定性随成分比而变化的规律.
团簇计算是在阿姆斯特丹密度泛函程序(ADF,版本号为2007.01)下完成的.这套计算程序对电荷密度采用广义梯度近似(GGA),对交换能的局域描写采用了Becke的梯度修正[25-26].在计算中,电子轨道用Slater型函数描述,并对砷原子和镓原子中直到3d轨道的内层电子均做冻结核近似处理.
2 结果及讨论
2.1 Ga12-n Asn(n=0~12)系列各团簇的基态结构
图1给出了Ga12-nAsn(n=0~12)系列团簇在基态时的结构图.从图1中可见,Ga12-nAsn(n=0~12)团簇的基态结构,从n=12时(12个砷原子)构成的一个两边各带一个帽的变形六边形多面体结构演变为n=0时(12个镓原子)构成的一个带有芯原子的多面菱形结构.特别是,对于砷和镓原子数目对等的Ga6As6结构,基态结构是带两个Ga-As帽的扭变棱柱结构,这个结构与1999年Yi用Car-Parrinello方法得到的结果相一致.在表1中,给出了Ga12-nAsn(n=0~12)系列团簇基态结构中各原子的空间坐标.
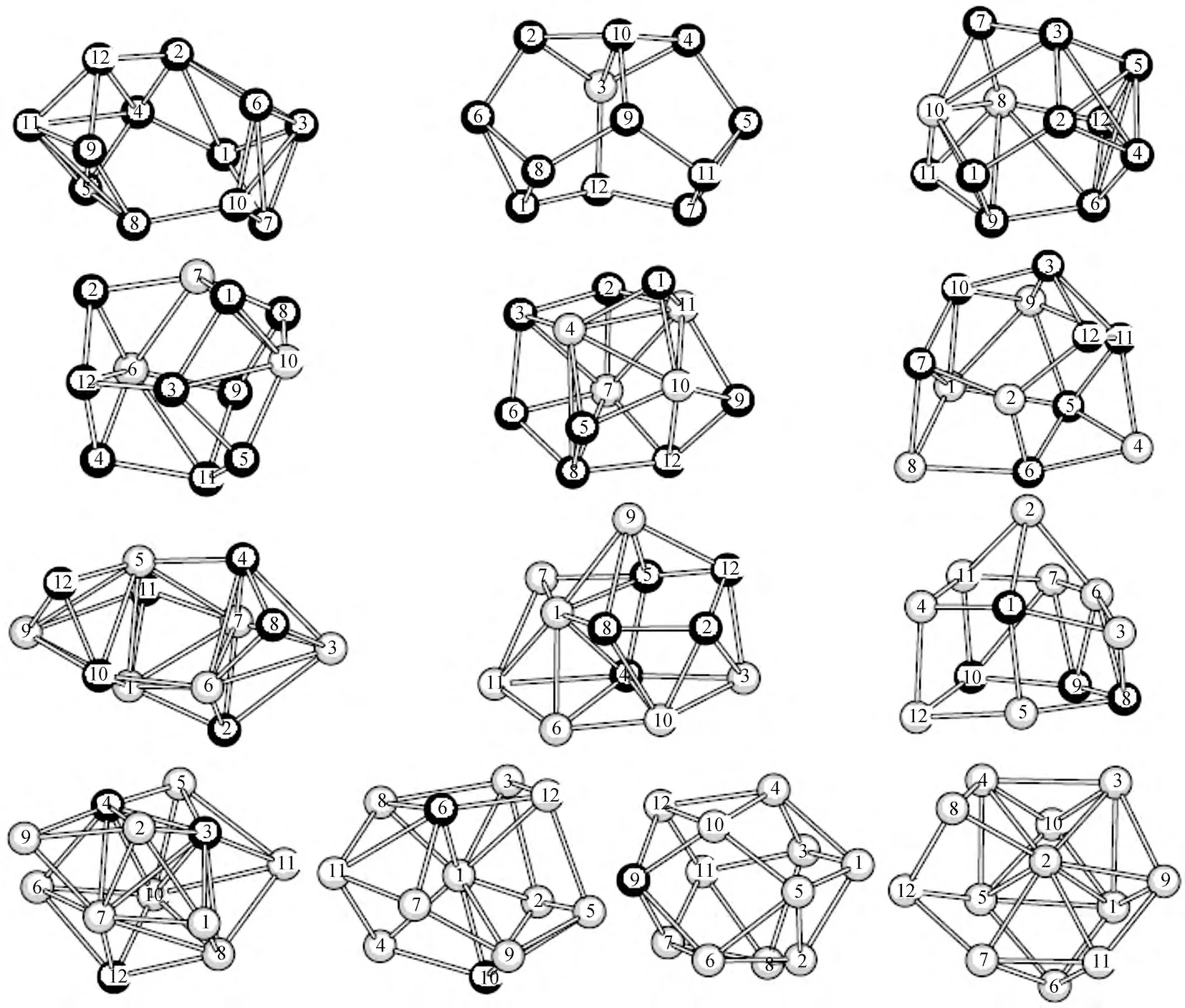
图1 Ga12-n Asn(n=0~12)团簇基态结构Fig.1 The structure of ground state of Ga12-n Asn(n=0~12)clusters
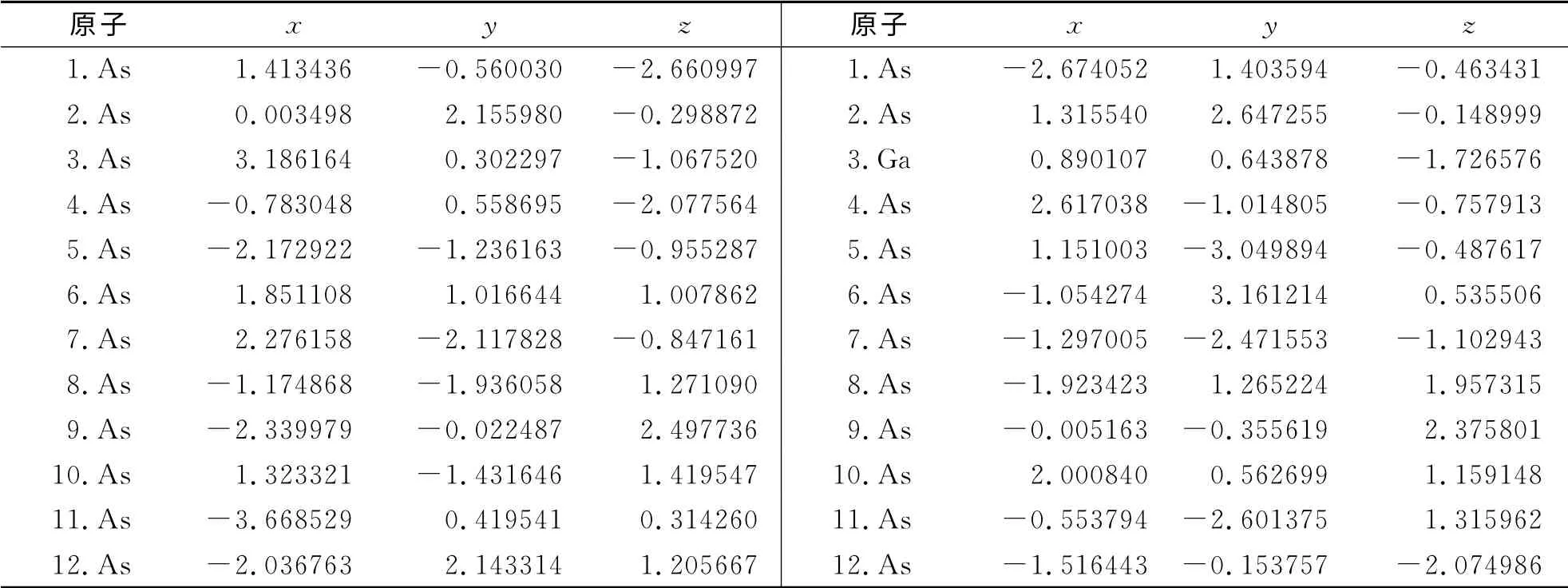
表1 Ga12-n Asn(n=0~12)团簇的基态结构中各原子的坐标参数Tab.1 The coordinate parameters of atoms of ground state structure of Ga12-n Asn(n=0~12)clusters /10-10 m
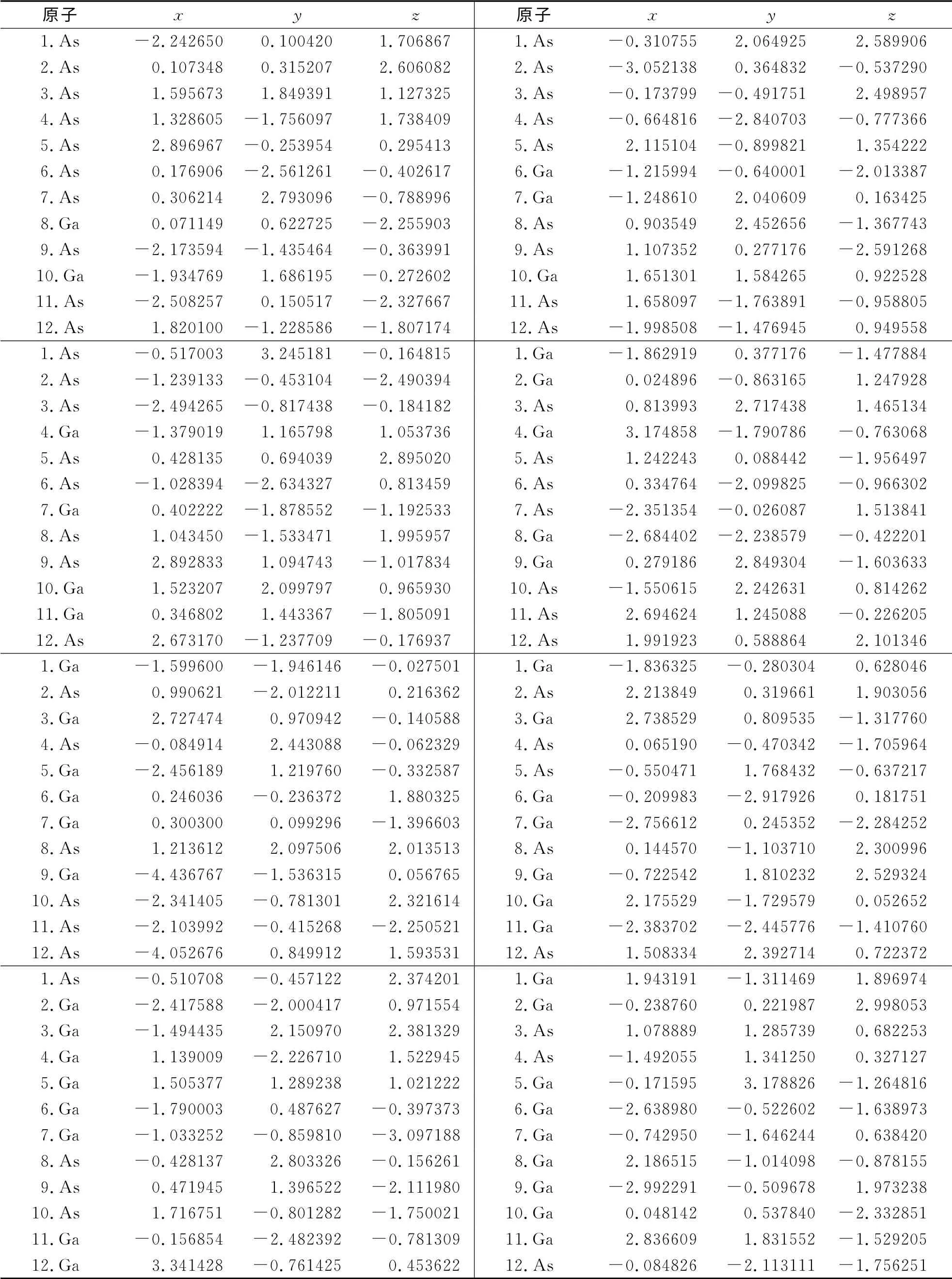
续表
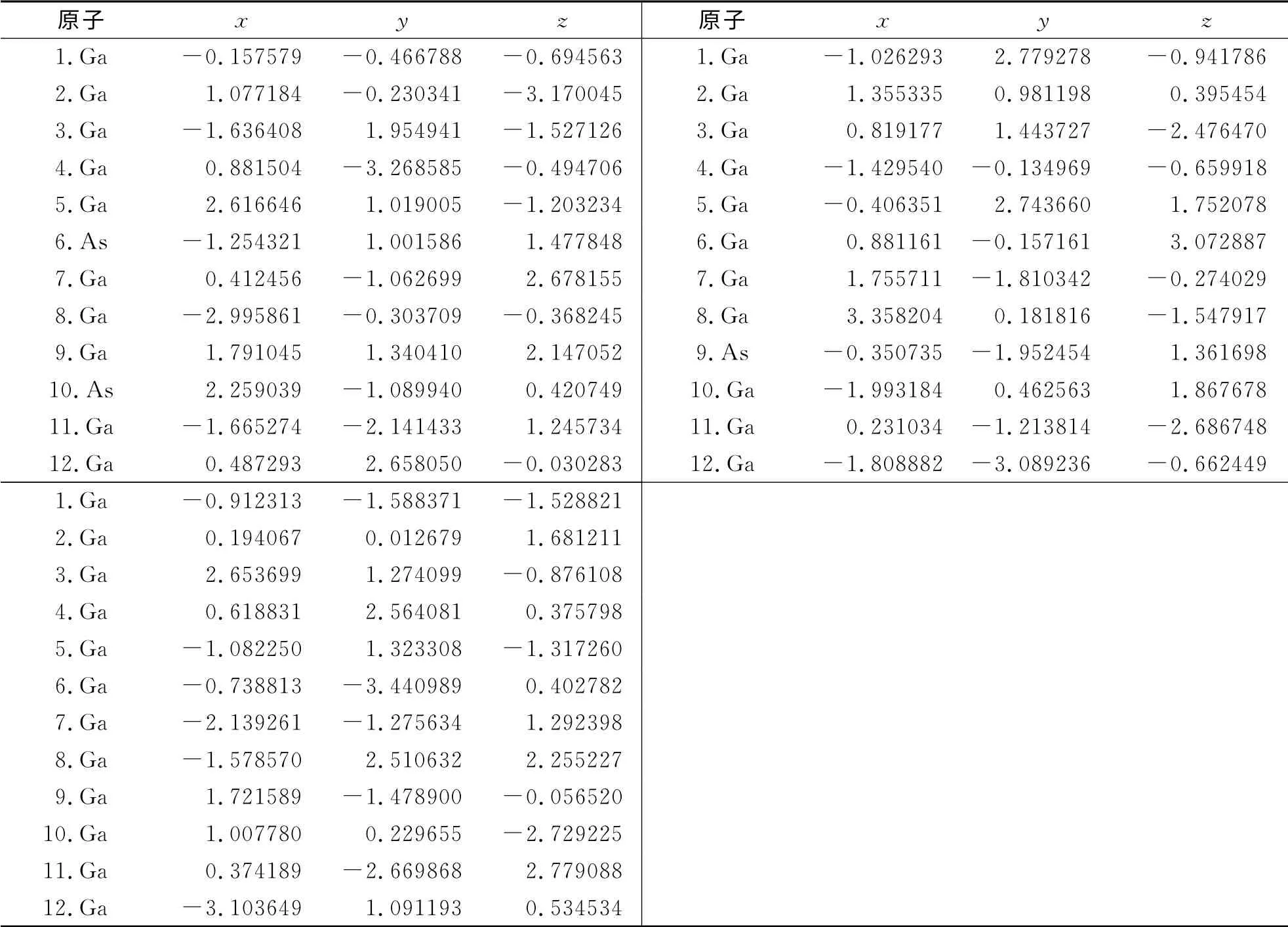
续表
2.2 Ga12-n Asn(n=0~12)团簇的能量和其他物理性质
表2 给出了Ga12-nAsn(n=0~12)团簇的总能量、电离能、亲和势、LDA 键能、交换关联能以及HOMO-LUMO 能隙.
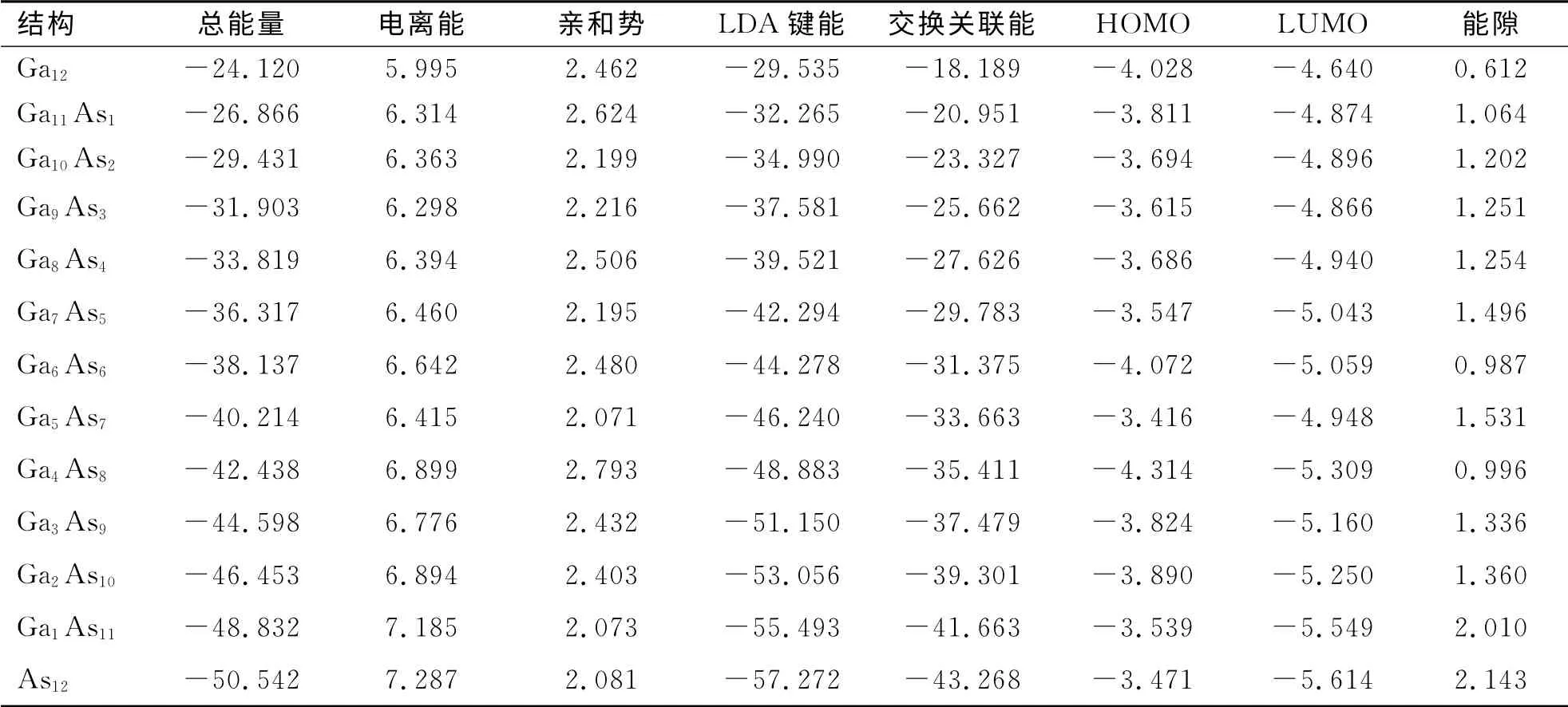
表2 Ga12-n Asn 的基态结构团簇的物理性质Tab.2 Physical properties of ground state structure of Ga12-n Asn(n=0~12)series clusters /eV
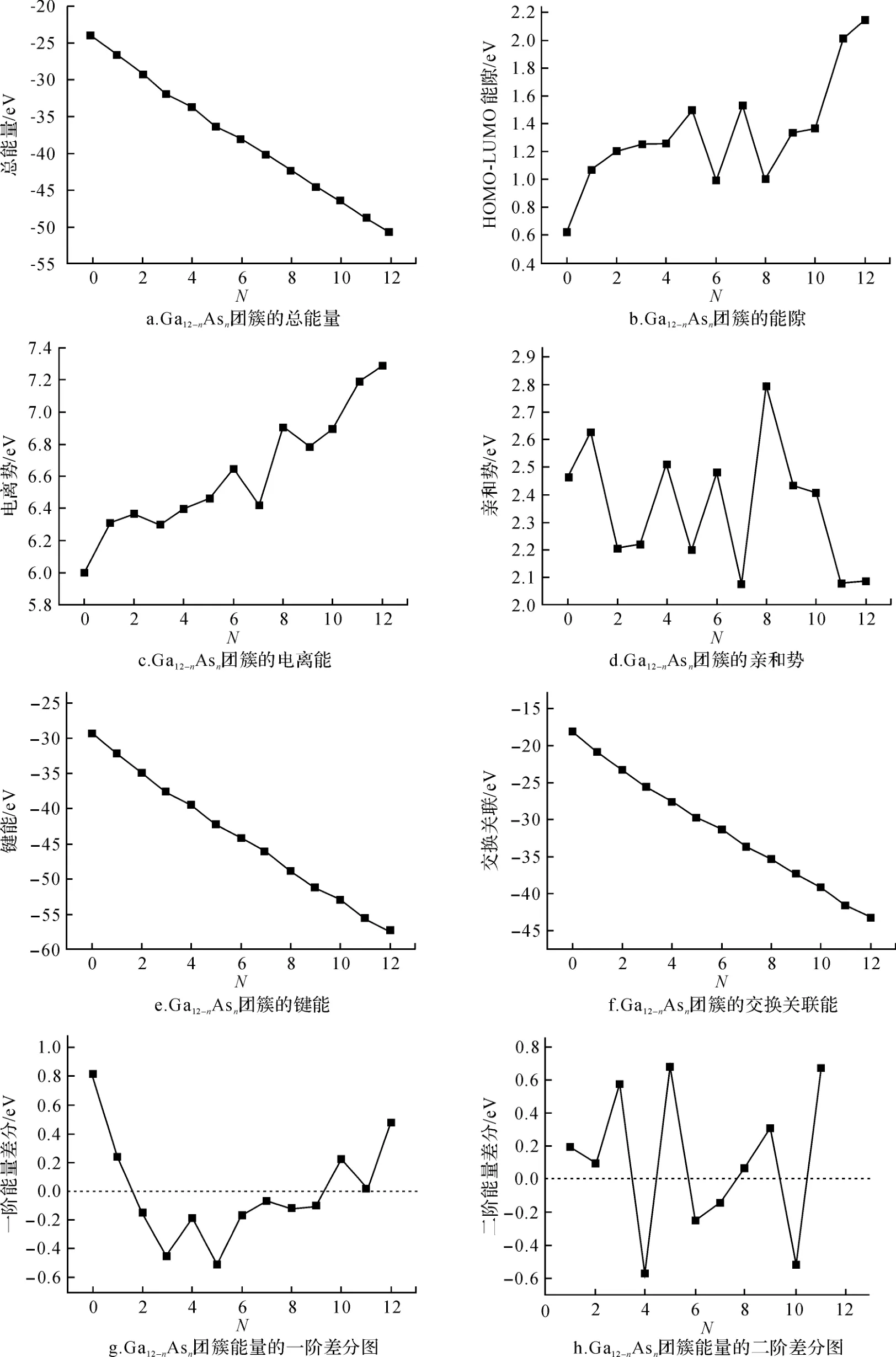
图2 Ga12-n Asn(n=0~12)基态结构团簇的总能量、HOMO-LUMO能隙、电离能等物理性质Fig.2 Total-energy,HOMO-LUMO gap,ionization-energy and other physical properties of ground state structure of Ga12-n Asn(n=0~12)series clusters
从表2 中可见,随着镓原子逐渐被砷原子取代,团簇的总能量、LDA 键能和交换关联能呈几乎线性的下降,亲和能表现出小幅的波动,电离能从5.9 e V 上升到7.3 e V,而HOMO-LUMO 能隙总体呈现出一种起伏有升的态势.在n=4~9之间,电离能特别是HOMO-LUMO 能隙表现出明显的奇偶性振荡.它们均在n=3、5、11处出现峰值,说明砷原子数目为这些值时,团簇的稳定性比较高.
从图2a中可见,随着砷原子个数的增加,团簇的总能量呈现几乎线性的下降(即结合能呈线性的增加).这也印证了一种普遍的观点[20],即原子数恒定的团簇中,含砷原子多的结构其结合能总会大一些.可以将这个变化规律用origin软件拟合为一个线性化的公式:

其中,斜率a=-2.17401,截距b=-24.93055.我们计算了各团簇能量与拟合值的差异,结果见图2g的能量一阶差分图.同时也计算了团簇能量的二阶差分值(即E(Ga12-n-1Asn+1)+E(Ga12-n+1Asn-1)-2E(Ga12-nAsn)),结果见图2h.从数据拟合中可见,在总的原子数保持为12时,用一个砷原子取代团簇中的镓原子,总能量将平均下降2.174 e V,或结合能将增加2.174 e V.从图2g中可见,当n=3和5时,结合能的一阶差分值出现高峰(总能量的一阶差分图出现低谷),将这个特点与图2b和图2h的特点相结合,我们可以说,在Ga12-nAsn(n=0~12)系列团簇中当n=3和n=5时稳定性将最高.
从图2c可见,随n的增加,电离能从5.9 e V 上升到7.3 e V,而图2 d中的亲和能在2.0~2.8 e V 之间波动,并明显小于电离能,这一方面说明Ga12-nAsn(n=0~12)团簇吸附一个电子要比丢失一个电子容易得多,另一方面也表明,对n比较大的团簇,即砷原子比较多的团簇,丢失电子而发生电离是比较困难的.
3 结论
本文在基于第一性原理的ADF程序(2007.01)平台上,对Ga12-nAsn(n=0~12)系列团簇的基态结构进行了大范围的随机筛选,并对筛选所得的低能量结构进行了精确计算,得到了Ga12-nAsn团簇各基态结构的总能量、HOMO-LUMO 能隙以及电离能、亲和势等物理性质.计算及分析表明,随着砷原子数目n的增加,团簇的总能量呈几乎线性的下降,在总原子个数不变的前提下,用一个砷原子去取代Ga12-nAsn团簇中的一个镓原子,团簇的总能量将平均下降2.174 eV.随着团簇中砷原子数目n的增加,LUMOHOMO 能隙从0.6 e V 上升到2.2 e V,并在n=4~9之间,呈现明显的奇偶性振荡.电离能从5.9 e V 上升到7.3 eV,亲和能却在2.0~2.7 eV 之间波动,电离能明显大于亲和能,说明中性团簇吸附一个电子比丢失一个电子来得容易.在HOMO-LUMO 能隙、总能量的一阶差分图和二阶差分图上均可看出,当n=3、5和11时团簇的稳定性比较高.
致谢 感谢丁望峰博士和李宝兴教授在本论文研究工作中给予的指导和帮助.
[1]Howes M J,Morgan D V.Gallium arsenate:materials,devices,and circuits[M].New York:Wiley,1986.
[2]O'Brien S C,Liu Y,Smalley R E,etal.Supersonic cluster beams of III-V semiconductors:GaxAsy[J].J Chem Phys,1986,84(7):4074-4079.
[3]Mohammad Al-Laham A,Raghavachari K.Theoretical study of Ga4As4,Al4P4,and Mg4S4clusters[J].J Chem Phys,1993,98(11):8770-8776.
[4]Song K M,Ray A K,Khowash P K.On the electronic structures of Ga As clusters[J].J Phys B,1994,27(8):1637-1648.
[5]Lou L,Wang L,Chibante L P F,etal.Electronic structure of small GaAs clusters[J].J Chem Phys,1991,94(38):8015-8020.
[6]Yi J Y.Atomic and electronic structures of small Ga As clusters[J].Chem Phys Lett,2000,325(1/2/3):269-274.
[7]Zhao J J,Xie R H,Zhou X L,etal.Formation of stable fullerenelike GanAsnclusters(6≤n≤9)Gradient-corrected density-functional theory and a genetic global optimization approach[J].Phys Rev B,2006,74(3):035319.
[8]Karamanis P,BéguéD,Pouchan C.Ab initio finite field(hyper)polarizability computations on stoichiometric gallium arsenide clusters GanAsn(n=2-9)[J].J Chem Phys,2007,127(9):75-77.
[9]Zhao W,Cao P L,Li B X,etal.Study of the stable structures of Ga4As4cluster using FP-LMTO MD method[J].Phys Rev B,2000,62(24):17138-17143.
[10]Zhao W,Cao P L,Study of the stable structures of the Ga5As5cluster using the full-potential linear-muffin-tin-orbital molecular-dynamics method[J].J Phys Condens Matter,2002,14:33-44.
[11]Zhao W,Cao P L.Study of the stable structures of Ga6As6cluster using FP-LMTO MD method[J].Phys Lett A,2001,288(1):53-57.
[12]Zhao W,Cao P L,Duan W H .Study of structure characteristics of the Ga8As8cluster[J].Phys Lett A,2006,349:224-229.
[13]Liu Y,Zhang Q L,Curl R F,etal.Photodetachment and photofragmentation studies of semiconductor cluster anions[J].J Chem Phys,1986,85(12):7434-7441.
[14]Taylor T R,Gomez H,Asmis K R,etal.Photoelectron Spectroscopy of GaX2-,Ga2X-,Ga2X2-,and Ga2X3-(X =P,As)[J].J Chem Phys,2001,115(10):4620-4631.
[15]Zhu X.Spectroscopic properties of gallium arsenide tetramers:Ga2As2,Ga2As2+and Ga2As2-[J].Spectrochimica Acta Part A,2005,61(11/12):2730-2736.
[16]Gutsev G L,Johnson E,Mochena M D,etal.The structure and energetics of(GaAs)n,(GaAs)n,and(Ga As)n+(n=2-15)[J].J Chem Phys,2008,128(14):144707.
[17]Gutsev G L,O'Neal Jr.R H,Saha B C,etal.Optical properties of(GaAs)nclusters(n=2-16)[J].J Phys Chem A,2008,112(43):10728-10735.
[18]Yang J,Li B,Zhan S.Study of GaAs cluster ions using FP-LMTO MD method[J].Phys Lett A,2006,348:416-423.
[19]杨建宋.带电对Ga7As7团簇基态结构的影响[J].杭州师范大学学报:自然科学版,2012,11(6):545-551.
[20]Liao M Z,Dai D G,Balasubramanian K.Electronic states of the Ga3As2and Ga2As3clusters[J].Chem Phys Lett,1995,239:124-130.
[21]马德明,李恩玲,施卫,等.密度泛函理论对GamAsn团簇的结构及稳定性的研究[J].原子与分子物理学学报,2008,25(4):984-990.
[22]马德明,施卫,李恩玲,等.Ga2Asn离子团簇结构及光电子能谱研究[J].光学学报,2009,29(4):1032-1037.
[23]姜科,刘乔升,杨建宋.Ga9-nAsn(n=0~9)系列团簇能量和稳定性的研究[J].杭州师范大学学报:自然科学版,2013,12(6):517-523.
[24]Ding W F,Li B X,A first-principles study of AlnSim-nclusters(m=6,9,10;n≤m)[J].J Mol Struc:THEO,2009,897:129-138.
[25]Becke A D.Density-functional exchange-energy approximation with correct asymptotic behavior[J].Phys Rev A,1998,38:3098-3100.
[26]Perdew J P,Density-functional approximation for the correlation energy of the inhomogeneous electron gas[J].Phys Rev B,1986,33:8822-8824.
猜你喜欢
——以物质结构与性质模块“元素周期律”教学为例
化学教与学(2023年3期)2023-02-09 08:33:22
物理学报(2021年12期)2021-07-01 09:42:56
原子与分子物理学报(2021年2期)2021-03-29 07:30:54
数学物理学报(2020年6期)2021-01-14 01:00:46
经济数学(2020年4期)2020-01-15 13:18:57
读与写·上旬刊(2018年10期)2018-11-27 20:03:08
中学生数理化·中考版(2017年12期)2017-04-18 12:55:09
武汉工程大学学报(2016年1期)2016-04-07 02:01:11
化学教与学(2014年6期)2014-07-03 10:04:09
水土保持通报(2014年5期)2014-06-09 08:26:42
