
伊潘立酮的合成新工艺
2015-03-19 01:57戴颖萍戴立言王晓钟陈英奇
浙江大学学报(工学版) 2015年3期
戴颖萍,戴立言,王晓钟,陈英奇
(浙江大学 化学工程与生物工程学系,浙江 杭州310027)
伊潘立酮,化学名为4-[3-[4-(6-氟-1,2-苯并异噁唑-3-基)-1-哌啶基]丙氧基]-3-甲氧基苯乙酮,是最新开发的非典型抗精神病药,临床用于治疗成年人精神分裂症.伊潘立酮属于5-HT2/D2受体拮抗剂,对多巴胺D3受体有很高的亲和力,对肾上腺素α1受体、多巴胺D4受体、5-HT6和5-HT7具有适当的亲和力,对5-HT1A、多巴胺D1和组胺H1受体有较低的亲和力.临床短期、长期的安全实验结果显示,伊潘立酮比目前使用的抗精神病药物的效果好且副作用少,并且该药物对缓解疼痛具有一定的疗效,是一种安全有效的抗精神病药物[1].
目前对伊潘立酮的合成有很多报道,但对该药的全合 成 涉 及 不 是 很 多.徐 勤 耀[2]、Bjork 等[3]和Strupczewsk等[4-6]报道以4-羟基-3-甲氧基苯乙酮和1-溴-3-氯丙烷为原料,在碳酸钾存在下合成4-(3-氯丙氧基)-3-甲氧基苯乙酮,再与6-氟-3-(4-哌啶基)-1,2-苯并异噁唑发生N-烷基化反应得到伊潘立酮.由于采用1-溴-3-氯丙烷为原料,反应过程中有副产物生成,导致产品的后处理及纯化较为困难.后处理需要通过减压蒸馏得到目标化合物,影响产品的纯度和收率.
在前人研究的基础上,设计一条新的合成路线:以(2,4-二氟苯基)-4-哌啶基甲酮盐酸盐(1)[7]为原料,与盐酸羟胺反应制得(2,4-二氟苯基)-4-哌啶基甲酮肟盐酸盐(2),然后在强碱作用下闭环得到6-氟-3-(4-哌啶基)-1,2-苯并异噁唑(3),再与3-氯丙醇发生 N-烷基化反应,得到6-氟-3-[1-(3-羟丙基)-4-哌啶基]-1,2-苯并异噁唑(4),最后与4-羟基-3-甲氧基苯乙酮(5)发生 Mitsunobu反应,制得伊潘立酮(8).该合成路线采用价格低、毒性低的3-氯丙醇代替1-溴-3-氯丙烷,避免副产物(7)的生成,得到高纯度的产品伊潘立酮,而且反应条件温和,收率良好,有较好的工业应用价值.同时,通过考察不同参数对反应的影响,确定合适的工艺条件.
1 试验部分
1.1 试验原料及仪器
各原料及其质量分数为:(2,4-二氟苯基)-4-哌啶基甲酮盐酸盐(自制),3-氯丙醇(99.5%,国药集团化学试剂有限公司),盐酸羟胺(98.5%,国药集团化学试剂有限公司),4-羟基-3-甲氧基苯乙酮(98.0%,阿拉丁试剂有限公司),三苯基膦(99.0%,阿拉丁试剂有限公司),偶氮二甲酸二异丙酯(DIAD)(95.0%,阿拉丁试剂有限公司),碳酸钾、氢氧化钠、无水硫酸镁等均为市售分析纯.
AVANCE DMX 500型核磁共振仪,Agilent 1200型高效液相色谱仪,GC 6820型气相色谱仪,Electrothermal IA 9200型熔点仪.
具体合成路线如图1所示.

图1 伊潘立酮的新合成路线Fig.1 New synthetic route of iloperidone
1.2 合成方法
1.2.1 (2,4-二氟苯基)-4-哌啶基甲酮肟盐酸盐(2)将 (2,4-二 氟 苯 基)-4-哌啶 基 甲 酮 盐 酸 盐 (1)(20.0 g,76.4 mmol)、盐 酸 羟 胺 (6.9 g,99.4 mmol)、100 m L无水乙醇加入到250 m L三口烧瓶中,加热至回流.薄层色谱(TLC)检测10 h后反应完全,停止加热,冷却至室温后析出白色固体,将反应液过滤,滤饼干燥后得17.5 g白色固体(2),收率为83.0%,高效液相色谱(HPLC)检测的产品纯度值为98.5%.熔点(mp)为258.6~258.9℃.(陆学华等[8]提到的熔点为256~258℃).
1.2.2 6-氟-3-(4-哌啶基)-1,2-苯并异噁唑(3) 将化合物(2)(15.0 g,54.2 mmol)、氢氧化钠(4.8 g,119.3 mmol)、90 m L水加入到250 m L三口烧瓶中,加热至回流反应1 h.TLC检测原料消失,停止反应,反应液冷却至室温,析出白色晶体,滤出晶体,水洗,真空干燥后得11.3 g白色固体(3),收率为94.5%,HPLC检测的产品纯度值为99.1%.mp为118.6~119.8℃.
1H NMR (500 MHz,CDCl3):δ7.68~7.71(dd,J1=5.0 Hz,J2=9.0 Hz,1H,Ar H),7.23~7.24(d,J=2.0 Hz,1H,Ar H),7.04~7.07(t,J=10.0 Hz,1H,Ar H),3.88(s,1H,NH),3.23~3.26(t,J=9.0 Hz,2H,CH2),2.79~2.85(t,J=13.5 Hz,2H,CH2),2.18 (m,1H,CH),2.04~2.06(q,J=10.5 Hz,2H,CH2),1.90~1.98(q,J=12.0 Hz,2H,CH2).
1.2.3 6-氟-3-[1-(3-羟丙基)-4-哌啶基]-1,2-苯并异噁唑(4) 将化合物(3)(10.0 g,45.4 mmol),3-氯丙醇 (4.7 g,49.9 mmol),K2CO3(6.3 g,45.4 mmol),KI(0.7 g,4.5 mmol),50 m L乙腈加入到100 m L三口烧瓶中,回流反应24 h.TLC检测原料基本消失,停止加热,冷却至室温,反应液减压浓缩后,加入50 m L水,用二氯甲烷萃取(50 m L×2),合并有机层,无水硫酸镁干燥,浓缩溶剂至干,用乙醇重结晶,得10.0 g白色晶体(4),收率为79.1%,HPLC(99.5%).mp为140.5~141.5℃.Mutlib等[9]提到的熔点为140~142℃)
1H NMR(500 MHz,CDCl3):δ7.69~7.74(dd,J1=5.5 Hz,J2=9.5 Hz,1 H,Ar H),7.24~7.26(d,J=2.0 Hz,1H,Ar H),7.05~7.10(t,J=9.0 Hz,1 H,Ar H),3.85~3.87(t,J=5.0 Hz,2H,CH2),3.31(s,1H,OH),3.25~3.29(t,J=9.0 Hz,2 H,CH2),2.81~2.83(t,J=12.5 Hz,2H,CH2),2.20(m,1H,CH),2.12~2.28(m,6 H,CH2),1.85~1.88(q,J=12.0 Hz,2H,CH2).
1.2.4 4-[3-[4-(6-氟-1,2-苯并异噁唑-3-基)-1-哌啶基]丙氧基]-3-甲氧基苯乙酮(8) 在N2保护下,将化合物(4)(1.0 g,3.6 mmol)、4-羟基-3-甲氧基苯乙酮(5)(0.9 g,5.4 mmol)、三苯基膦(1.2 g,4.7 mmol)、10 m L无水四氢呋喃加入25 m L三口烧瓶中,冰浴条件下缓慢滴加DIAD(0.9 g,4.7 mmol),冰浴反应.TLC检测1 h后反应完全,反应液经柱层析分离,得到1.2 g白色固体伊潘立酮(8),收率为75.7%,HPLC(99.8%).mp为119.8~120.3℃ (Bordeau等[10]提到的熔点为118~120℃).
1H NMR(500 MHz,CDCl3):δ6.92~7.74(m,6H,Ar H),4.19~4.21(t,J=6.25 Hz,2 H,CH2),3.92(s,3 H,CH3),3.16(m,3 H,CH,CH2),2.71(m,2H,CH2),2.57(s,3H,CH3),2.31 (m,2 H,CH2),2.17 (m,6 H,CH2);HRMS calcd.for C24H27N2O4F 426.195 5,found 426.195 5.
2 试验结果与讨论
2.1 (2,4-二氟苯基)-4-哌啶基甲酮肟盐酸盐
2.1.1 盐酸羟胺用量对反应的影响 在肟化反应中,羟胺的用量都是过量的,过量大多为30%,有的高达100%[11].针对盐酸羟胺用量的试验,原料投料量均为(2,4-二氟苯基)-4-哌啶基甲酮盐酸盐20.0 g(76.4 mmol),乙醇100 m L,回流反应10 h.不同盐酸羟胺用量(mA)下所得反应收率(Y)如表1所示.
由表1可知,随着盐酸羟胺用量的增加,反应收率明显增大.当盐酸羟胺与(2,4-二氟苯基)-4-哌啶基甲酮盐酸盐的物质的量的比为1.3:1时,反应收率达83.0%,再增加盐酸羟胺的用量对收率影响不大.故选用盐酸羟胺与(2,4-二氟苯基)-4-哌啶基甲酮盐酸盐的物质的量比为1.3:1作为反应配比.

表1 盐酸羟胺用量对肟化反应的影响Tab.1 Effect of hydroxylamine hydrochloride amount on_______oximation reaction
2.1.2 反应时间对反应的影响 反应时间是影响肟化反应的另一个重要因素,针对反应时间进行的试验,原料投料量均为(2,4-二氟苯基)-4-哌啶基甲酮盐酸盐20.0 g(76.4 mmol),盐酸羟胺6.9 g(99.4 mmol),乙醇100 m L,回流反应.不同反应时间下所得反应收率如表2所示.由表2可知,当反应时间达10 h时,再继续延长反应时间对收率影响不大,故最佳反应时间选择10 h.

表2 反应时间对肟化反应的影响_Tab.2 Effect of reaction time on oximation reaction
2.2 6-氟-3-(4-哌啶基)-1,2-苯并异噁唑
闭环反应在强碱作用下进行,试验发现氢氧化钾和氢氧化钠对反应都有较好的效果,本研究选用氢氧化钠进行反应.通过改变氢氧化钠用量,考察碱的用量对反应的影响,原料投料量均为(2,4-二氟苯基)-4-哌啶基甲酮肟盐酸盐15.0 g(54.2 mmol),氢氧化钠质量分数保持5%,回流反应1 h.在不同氢氧化钠用量(mC)下所得反应收率如表3所示.由表3可知,当氢氧化钠的用量为2.2当量时,再继续增加氢氧化钠的用量对收率几乎没影响,故选择2.2当量的氢氧化钠.

表3 氢氧化钠用量对闭环反应的影响Tab.3 Effect of sodium hydroxide amount on ring closure_______reaction
2.3 6-氟-3-[1-(3-羟丙基)-4-哌啶基]-1,2-苯并异噁唑(4)
2.3.1 溶剂对反应的影响 N-烷基化反应一般在极性溶剂中进行.针对几种常用极性溶剂所进行的试验,原料投料量均为6-氟-3-(4-哌啶基)-1,2-苯并异噁唑10.0 g(45.4 mmol),3-氯丙醇4.7 g(49.9 mmol),K2CO36.3 g(45.4 mmol),KI 0.7 g(4.5 mmol),回流反应.在不同溶剂中,不同温度(θ)和反
应时间(t)下所得反应收率如表4所示.
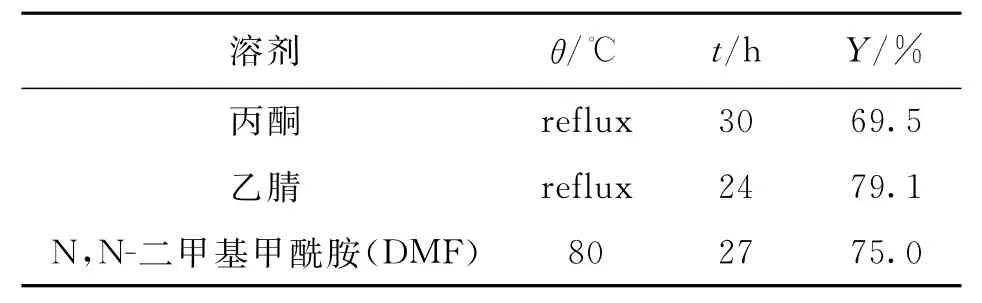
表4 反应溶剂对N-烷基化反应的影响___Tab.4 Effect of solvents on N alkylation reaction
由表4可知,用丙酮作溶剂,反应收率比乙腈和DMF较低,因为丙酮的回流温度为56℃,反应温度低导致反应不完全,收率较低;如果N-烷基化反应的温度太高,3-氯丙醇自身易发生醚化副反应,所以在用DMF作溶剂时选择反应温度为80℃,收率和在乙腈回流温度下反应接近,考虑到DMF沸点较高,工业化操作比较困难,故选择乙腈作为反应溶剂.
2.3.2 3-氯丙醇用量对反应的影响 3-氯丙醇作为烷化剂,反应效果较好且价格低廉.针对3-氯丙醇用量进行的试验,原料投料量均为6-氟-3-(4-哌啶 基 )-1,2-苯 并 异 噁 唑 10.0 g (45.4 mmol),K2CO36.3 g(45.4 mmol),KI 0.7 g(4.5 mmol),50 m L乙腈,回流反应24 h.不同3-氯丙醇用量(mD)下所得反应收率如表5所示.由表5可知,当3-氯丙醇用量为1.1当量时,收率达到79.1%,再增加3-氯丙醇的量对收率影响不大,故选择3-氯丙醇与6-氟-3-(4-哌啶基)-1,2-苯并异噁唑的物质的量的比为1.1:1为反应配比.

表5 3-氯丙醇用量对N-烷基化反应的影响Tab.5 Effect of 3-chloropropanol amount on N alkylation______reaction
2.3.3 催化剂对反应的影响 Bolos[12]和刘员等[13]提到类似的反应,选用溴化钠或碘化钾为催化剂可提高3-氯丙醇的反应活性,以促进该类反应进行,提高反应收率,故对催化剂进行考察.原料投料量均为 6-氟-3-(4-哌 啶 基)-1,2-苯 并 异 噁 唑 10.0 g(45.4 mmol),3-氯丙醇4.7 g(49.9 mmol),K2CO36.3 g(45.4 mmol),50 mL乙腈,回流反应24 h.不同催化剂及其用量(mE)下所得反应收率如表6所示.
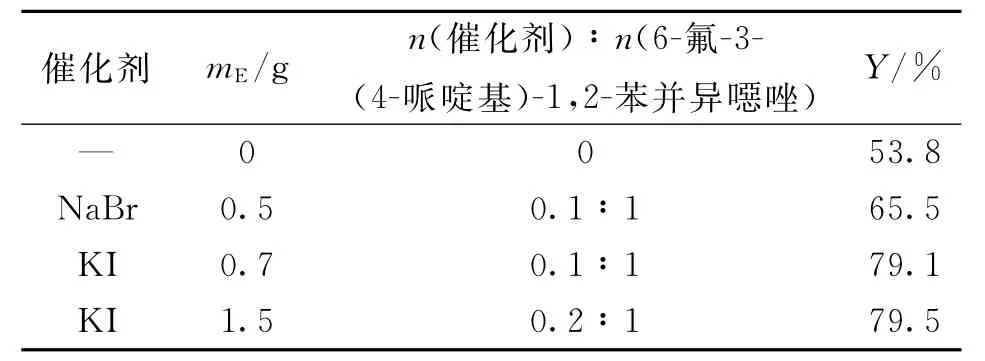
表6 催化剂N-烷基化对反应的影响___Tab.6 Effect of catalysts on N alkylation reaction
由表6可见,催化剂对反应收率的影响非常明显,碘化钾的催化效果比溴化钠好.当碘化钾催化量由0.1当量增加到0.2当量时,反应收率增加不明显,故选用0.1当量的碘化钾作为催化剂.
2.4 4-[3-[4-(6-氟-1,2-苯并异噁唑-3-基)-1-哌啶基]丙氧基]-3-甲氧基苯乙酮(8)
2.4.1 4-羟基-3-甲氧基苯乙酮用量对反应的影响
在Mitsunobu反应中,醇与带有活性氢的各种亲核试剂进行脱水缩合反应,构建C-O、C-S、C-N、C-C等键.亲核试剂一般是带有-OH、-SH、-NH等基团的酸性化合物,其酸度系数p Ka≤15.常见的亲核试剂有羧酸、酚、酰亚胺、嘌呤、硫醇等.4-羟基-3-甲氧基苯乙酮(p Ka<9)作为亲核试剂,其用量对于反应有重要影响.针对亲核试剂用量的试验,原料投料量均为6-氟-3-[1-(3-羟丙基)-4-哌啶基]-1,2-苯并异噁 唑 1.0 g (3.6 mmol),Ph3P 1.2 g (4.7 mmol),DIAD 0.9 g(4.7 mmol),10 m L无水四氢呋喃,冰浴反应1 h.在不同亲核试剂用量(mF)下所得反应收率如表7所示.由表7可知,当亲核试剂的用量达到1.5当量时,反应已完全,收率达75.7%,再增加用量对收率影响不大,故选择4-羟基-3-甲氧基苯乙酮与6-氟-3-[1-(3-羟丙基)-4-哌啶基]-1,2-苯并异噁唑的物质的量的比为1.5:1为反应配比.

表7 4-羟基-3-甲氧基苯乙酮用量对Mitsunobu反应的影响Tab.7 Effect of 4-hydroxy-3-methoxyacetophenone amount______on Mitsunobu reaction
2.4.2 Mitsunobu试剂用量对反应的影响 对于亲核试剂的p Ka<11的 Mitsunobu反应来说,Ph3P-DIAD体系作为反应试剂能得到较高的收率.针对Mitsunobu试剂用量的试验,原料投料量均为6-氟-3-[1-(3-羟丙基)-4-哌啶基]-1,2-苯并异噁唑1.0 g(3.6 mmol),4-羟基-3-甲氧基苯乙酮0.9 g(5.4 mmol),10 m L无水四氢呋喃,冰浴反应1 h.不同Ph3P用量(mG)和DIAD用量(mH)下所得反应收率如表8所示.由表8可知,当Ph3P、DIAD和6-氟-3-[1-(3-羟丙基)-4-哌啶基]-1,2-苯并异噁唑的物质的量之比为1.3:1.3:1时,反应已基本完全,收率达75.7%,再增加Mitsunobu试剂用量对收率基本没影响,故选择Mitsunobu试剂的物质的量为1.3当量.
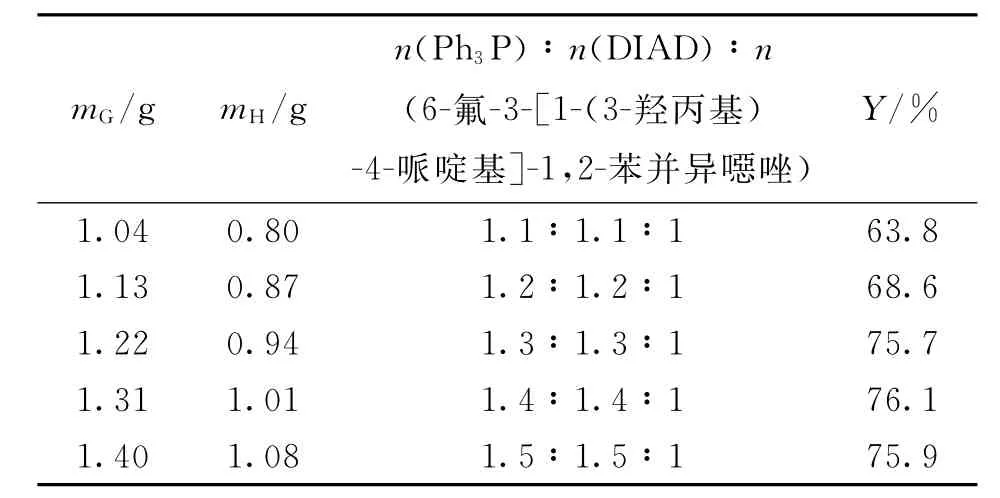
表8 Mitsunobu试剂用量对Mitsunobu反应的影响Tab.8 Effect of Mitsunobu reagent amount on Mitsunobu_______reaction
3 结 论
(1)以(2,4-二氟苯基)-4-哌啶基甲酮盐酸盐为原料,经过肟化、闭环、N-烷基化、Mitsunobu反应得到伊潘立酮,总收率达47.0%以上.该合成路线操作简便、收率良好,因此具有较好的工业应用前景.
(2)肟化反应一般要加碱(如碳酸钠、氢氧化钠)来中和盐酸羟胺,以释放游离的羟胺.试验发现,肟化反应不需要加碱,直接用盐酸羟胺与(2,4-二氟苯基)-4-哌啶基甲酮盐酸盐反应也能进行,同时优化反应条件,减少盐酸羟胺的用量以降低生产成本.
(3)环合反应在碱性水溶液中进行,反应结束后冷却,直接析出产品,无需溶剂萃取,不仅简化了试验操作,而且避免了有机溶剂的使用,路线更为绿色环保.
(4)在N-烷基化反应中,采用价格低、毒性低的3-氯丙醇代替1-溴-3-氯丙烷,与6-氟-3-(4-哌啶基)-1,2-苯并异噁唑反应.由于3-氯丙醇中2个取代基反应活性不同,羟基不易被取代,通过控制反应条件来防止其副产物的生成,得到纯度为99.8%的产物.
(5)在 Mitsunobu反应条件下,醇类化合物和酸性化合物易发生分子间脱水反应,形成C-O键.通过Mitsunobu反应来合成伊潘立酮,反应条件温和、产品纯度和收率都较高.
(
):
[1]朱玉莹,胡春.伊潘立酮[J].中国药物化学杂志,2009,19(5):400.ZHU Yu-ying,HU Chun.Iloperidone[J].Chinese Journal of Medicinal Chemistry,2009,19(5):400.
[2]徐勤耀,任白燕,胡文浩,等.伊潘立酮的合成[J].中国医药工业杂志,2011,42(2):88- 89.XU Qin-yao,REN Bai-yan,HU Wen-hao,et al.Synthesis of iloperidone[J].Chinese Journal of Pharmaceuticals,2011,42(2):88- 89.
[3]BJORK A K K,ABRAMO A L,KJELLBERG B E S.Aryl ethers of N-alkyl-piperidines and acid addition salts thereof:US,4,366,162[P].1982-12-28.
[4]STRUPCZEWSKI J T.Intermediate compounds in the synthesis of heteroarylpiperidines,pyrrolidines and piperazines:US,5,663,449[P].1997-09-02.
[5]STRUPCZEWSKI J T.N-(aryloxyalkyl)heteroarylpiperidines and-heteroarylpiperazines,a process for their preparation and their use as medicaments:EP,0,402,644[P].1995-08-16.
[6]STRUPCZEWSKI J T,ALLEN R C,GLAMKOWSKI E J,et al.Synthesis and neuroleptic activity of 3-(1-substituted-4-piperidinyl)-1,2-benzisoxazoles[J].Journal of Medicinal Chemistry,1985,28(6):761- 769.
[7]沈健芬,郑睿.2,4-二氟苯基-4-哌啶基甲酮盐酸盐的合成工艺研究[J].精细化工中间体,2010,40(4):34- 37.SHEN Jian-fen,ZHENG Rui.Research on the synthesis of(2,4-difluorophenyl)(piperidin-4-yl)methanone hydrochloride[J].Fine Chemical Intermediates,2010,40(4):34- 37.
[8]陆学华,潘莉,唐承卓,等.利培酮的合成[J].中国药物化学杂志,2007,17(2):89- 91.LU Xue-hua,PAN Li,TANG Cheng-zhuo,et al.Synthesis of risperidone[J].Chinese Journal of Medicinal Chemistry,2007,17(2):89- 91.
[9]MUTLIB A E,STRUPCZEWSKI J T,CHESSON S M.Application of hyphenated LC/NMR and LC/MS techniques in rapid identification of in vitro and in vivo metabolites of iloperidone [J].Drug Metabolism and Disposition,1995,23(9):951- 964.
[10]BORDEAU K J,CHIANG Y,STRUPCZEWSKI J T,et al.3-[[(aryloxy)alkyl]piperidinyl]-1,2-benzisoxazoles as D2/5-HT2antagonists with potential atypical antipsychotic activity:antipsychotic profile of iloperidone(HP 873)[J].Journal of Medicinal Chemistry,1995,38(7):1119- 1131.
[11]周玉林,金柏林.肟化、羟肟化反应中降低羟胺用量的方法[J].有色金属,1992,3:45- 46.ZHOU Yu-lin,JIN Bai-lin.The method of reducing hydroxylamine in oximation,hydroxyl oximation reactions[J].Nonferrous Metals,1992,3:45- 46.
[12]BOLOS J,GUBERT S,ANGLADA L,et al.7-[3-(1-piperidinyl)propoxy]chromenones as potential atypical antipsychotics[J].Journal of Medicinal Chemistry,1996,39(15):2962- 2970.
[13]刘员,南云,戴立言,等.3-正丙基-2,4-二羟基苯乙酮的合成工艺[J].浙江大学学报:工学版,2012,46(9):1697- 1701.LIU Yuan,NAN Yun,DAI Li-yan,et al.Synthesis technology of 3-n-propyl-2,4-dihydroxyacetophenone[J].Journal of Zhejiang University:Engineering Science,2012,46(9):1697- 1701.
猜你喜欢
分子催化(2022年1期)2022-11-02
健康体检与管理(2022年2期)2022-04-15
能源化工(2021年6期)2021-12-30
食品工业科技(2021年23期)2021-12-16
精神医学杂志(2021年2期)2021-06-23
中国医药指南(2019年30期)2019-11-28
消费导刊(2019年14期)2019-08-21
消费导刊(2019年27期)2019-07-22
中国油脂(2017年7期)2017-09-16
教育教学论坛(2017年37期)2017-09-14
