
基于随机游走的蛋白质功能预测算法设计与实现
2015-03-17 11:56:02马吉权贾翠翠张军杰
黑龙江大学工程学报 2015年3期
马吉权, 贾翠翠,张军杰
(1.黑龙江大学 计算机科学技术学院,哈尔滨 150080;2.山东航天电子技术研究所,山东 烟台 264670)
基于随机游走的蛋白质功能预测算法设计与实现
马吉权1, 贾翠翠1,张军杰2
(1.黑龙江大学 计算机科学技术学院,哈尔滨 150080;2.山东航天电子技术研究所,山东 烟台 264670)
在构建蛋白质相互作用网络时从通路数据出发, 利用通路数据整合蛋白质相互作用网络。提出了一个基于随机游走的蛋白质功能预测方法, 在该算法中把已知功能的蛋白质当作起始点, 对于随机游走在蛋白质互作网络中产生的邻居信息, 转换为注释模式信息, 并根据已知功能的蛋白质的功能来提取未知功能蛋白质的注释模式。利用传统的K近邻算法从训练样本集中找到未知功能蛋白质的k个最近邻。最后, 结合多标签分类的K近邻算法, 统计k个近邻中蛋白质的功能类数目, 基于最大后验概率预测未知功能蛋白质属于的功能标签类。通过在构建蛋白质互作网络进行实验, 结果表明提出的方案能够有效地进行蛋白质功能预测。
蛋白质功能预测;蛋白质-蛋白质相互作用网络;随机游走;蛋白质功能注释;K近邻;多标签分类
0 引 言
近年来,基于高通量技术酵母双杂交[1]实验, 大量的蛋白质相互作用网络被构建并用于蛋白质功能的预测。一般认为, 蛋白质相互作用网络中相邻的蛋白质表现类似的功能, 提出了很多基于蛋白质相互作用网络的蛋白质功能预测方法[2],比如多标签分类算法[3]、半监督学习[4]等方法。也有学者提出了基于随机游走的蛋白质功能预测方法[5], 解决了相互作用数据的功能分布和相互作用网络的全局拓扑结构问题。随机游走算法综合了网络的全局和局部拓扑结构信息, 度量未知功能的蛋白质和网络中其他蛋白质的关联程度。
到目前, 人们通过实验方法已经确定了蛋白质间的一些相互作用, 根据这些相互作用建立了一些蛋白质相互作用数据库, 并了解相互作用的细节, 累积了一些蛋白质相互作用有关的专业知识。但是, 还不能够构建完整精确的蛋白质相互作用网络, 如何利用已有的数据预测出未知的蛋白质相互作用, 以及有效地分析蛋白质相互作用网络, 从而从更深层次了解蛋白质的功能, 成为蛋白质组学研究的一个重要问题。根据蛋白质相互作用和蛋白质功能预测问题, 通过整合一型糖尿病数据和BioGrid[6]数据库下载到的数据构建了一个蛋白质相互作用网络, 在相互作用网络上实现了随机游走算法, 结合K近邻算法实现了蛋白质功能的预测。
1 数据的获取与处理
尽管有许多预测蛋白质功能的方法,但大多数都只是使用了一种数据, 而随着研究的深入, 越来越多的人把其它数据信息融合到相互作用网络从而实现蛋白质的功能注释与预测。根据一型糖尿病通路数据构建一个蛋白质相互作用网络, 目的是让整个网络的功能都是相关的, 从而提高预测的准确性。
1.1 数据的获取
本文主要对一型糖尿病蛋白质进行分析, 并根据一型糖尿病数据构建了蛋白质相互作用网络。注释数据是从GO网站下载得到的所有物种的注释信息, 采用GO功能注释[7]格式对蛋白质进行功能注释。
Data 1: 来源于一型糖尿病的通路数据, 手工收集到了与一型糖尿病相关的202个蛋白质。
Data 2: 从BioGrid数据库下载得到的蛋白质相互作用数据, 其版本号为2.0.63。该数据集共包含336 168条蛋白质相互作用数据。BioGrid由酵母、线虫、果蝇以及人类蛋白质相互作用共4个子数据库组成。
1.2 数据的整合
笔者在下载到的BioGrid数据表中找到了122个一型糖尿病蛋白质相关的相互作用关系。在删除自身相互作用和重复数据后, 构建了蛋白质相互作用网络。发现在大的网络外还有一些小的相互作用关系, 在删除这些互作关系数据后,最终得到了蛋白质相互作用数据包括1 093个蛋白质和1 708条相互作用对。
笔者认为互作网络中数据源Data 1中的蛋白质是已知功能的蛋白质, 而数据源Data 2中的蛋白质是未知功能的, 在GO网站下载到了所有物种的功能注释文件, 对数据源Data 1中的蛋白质到GO下载到的注释文件中查找它们的功能并且对其功能进行了统计, 取出了它们共同拥有的功能最多的前16个。根据这些功能类, 对数据源Data 2中数据进行了处理, 留下有互作关系的两个蛋白质都至少有16个功能类中的一个互作关系, 并且对Data2中各个功能类中注释信息所包含的蛋白质数量进行了分析统计, 见图1。

图1 各个功能类所注释的蛋白质的数量Fig.1 Number of protein of function annotations
2 基于蛋白质相互作用网络的随机游走算法的设计与实现
随机游走模型的思想是[8]: 粒子从一个或若干个顶点开始游走一张图, 从任意一个起始点开始, 以一定的概率pt通过边随机的移动到这个顶点的邻居顶点上, 每次游走后得出一个概率分布pt+1,pt+1体现了图中起始点随机的移动到与它相邻的各顶点的概率, 然后把pt+1的值赋给pt当作下一次随机移动的输入并且重复执行这个经过。在符合特殊的条件时,pt与pt+1的变化值会趋于收敛的状态, 在这个状态下得到一个稳定的概率分布。
从形式上看, 随机游走重新启动被定义为:
(1)
其中,W是列规范化图的邻接矩阵, 表示蛋白质相互作用网络的相互作用关系, 矩阵W中的元素wij表示蛋白质i和j的相互作用关系,d(V)表示与蛋白质i有相互作用的蛋白质的个数。wij=1/d(V)表示蛋白质i和j间存在某种相互作用关系,wij=0表示蛋白质i和j间没有互作关系。
用W=(wij),i,j∈V表示邻接矩阵,wij代表W中的每一个元素, 其中
(2)
r表示粒子每次游走时回到初始位置的概率。越接近 0, 随机游走过程越能体现围绕起始点的网络全局拓扑特性, 值越接近1, 则越能体现网络局部结构性质。p0为n×1的向量, 表示粒子随机游走的初始状态,将网络中已知功能的蛋白质的初始值设为相等的值, 并且所有这些值的和为1, 其余未知功能的蛋白质均为0。pt是一个向量, 它保存节点i在t时刻的概率。
pt+1是经过第t+1次游走后,粒子在各个蛋白质处出现的概率。经过多次游走, 粒子在各蛋白质处出现的概率将趋于稳定, 根据在稳定状态概率向量p∞的值分别排列未注释功能的蛋白质。p∞的值是在向量pt和向量pt+1之间的差距小于10-6时获得的。p∞与蛋白质相互作用网络的拓扑结构紧密相关, 能够度量已知功能的蛋白质与相互作用网络中未知功能的蛋白质间的关系, 称稳态分布下概率值为起始蛋白质的全局邻居或邻居模式。
3 基于邻居模式的蛋白质功能预测
传统的预测蛋白质功能的方法都是假设:在相互作用网络中拓扑距离较近的蛋白质更倾向于具有相似的功能[9]。并没有利用到蛋白质的注释信息来对蛋白质进行功能注释, 因此, 假设功能相似的蛋白质具有相似的注释模式, 充分考量了蛋白质注释模式的相似性对蛋白质功能注释的影响。在蛋白质相互作用网络中, 考虑的范围不仅是相邻的蛋白质, 而是整个网络[10], 通过与已经注释的蛋白质的注释模式进行比较来预测未知蛋白质的功能。如果两个蛋白质的注释模式相似, 则这两个蛋白质就具有相似的功能。
提取注释模式可分为:①在构建的蛋白质相互作用网络上执行随机游走算法, 经过多次迭代后得到最终的稳态向量, 即目标节点的全局邻居;②把相互作用网络的特征向量, 转换为基于功能的特征向量, 也就是注释模式。
在互作网络中蛋白质的功能与其相邻的蛋白质的功能是相似的, 因此, 用执行随机游走算法后得到的稳态向量来表示蛋白质的注释环境Si,i∈[1,n], 即
(3)
表示以i点为起始点, 执行随机游走算法后得到稳态向量后, 蛋白质i到全局网络其他蛋白质的亲和向量。
相互作用网络见图2, 白色的圆表示未知功能的蛋白质, 蓝色的圆表示已知功能的蛋白质, 方框中为蛋白质所具有的功能, 边上的权值Sij表示节点i到节点j的相近度。在对未知功能蛋白质提取注释模式时, 根据与未知功能的蛋白质相邻的已知功能的蛋白质的功能来提该蛋白质的注释模式。
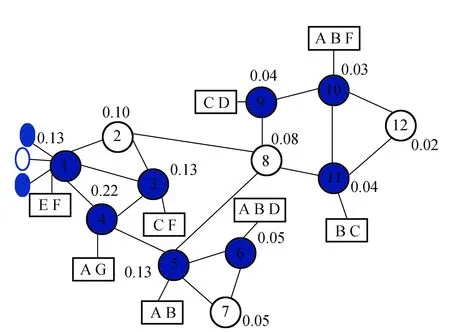
图2 执行随机游走算法后蛋白质相互作用网络示意图Fig.2 Diagram of the protein interaction network after random walk
图2是邻居模式转换为注释模式的过程, 图2为随机游走算法以蛋白质4为起始点执行的结果, 邻居模式为(0.13, 0.04, 0.04)分别对应(5, 9,11)号蛋白质, 蛋白质5具有功能A、B, 蛋白质11具有功能B、C, 蛋白质9具有功能C、D。通过对这些功能对应的权值进行求和, 得到向量(0.13, 0.17, 0.08, 0.04), 分别对应功能A,B,C,D。之后对得到的向量进行归一化, 得到蛋白质4的功能注释模式
在蛋白质i的功能注释模式中, 其中功能a所对应的概率值为:
(4)
当蛋白质j具有功能a时,eja的值为1, 否则为0。
所有的功能类对应的概率向量为:
(5)
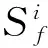
4 多标签学习的K近邻算法
该算法的思想是[11]:对于某一个测试样本, 首先在训练集中找到它的k个最近邻样本点[12], 然后根据训练样本集中的统计信息, 比如这k个最近邻样本属于任意标签的样本个数, 以及在训练过程中这k个最近邻样本属于任意标签的样本个数和中心样本点是不是含有该标签等, 把这些信息与条件概率知识及贝叶斯算法结合起来, 从而预测待测样本的标签集。
5 实验与结果分析
用已知功能的蛋白质组成了一个小网络, 这个网络用来验证使用。在这个小网络中一共有52个蛋白质, 取出1/7蛋白质作为未知的,用网络中其它的已知功能的蛋白质来预测这些蛋白质的功能。 这1/7蛋白质共有44个功能, 笔者首先实现了单标签K近邻算法, 验证结果见图3。
实现多标签的K近邻算法, 并将其结果与单标签的结果进行比较, 见图4。 由图4可见, 多标签的K近邻算法明显优于单标签的K近邻算法。
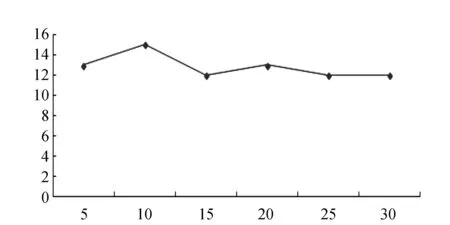
图3 不同k值预测出蛋白质功能的个数Fig.3 Numbers of different k value to predict protein function
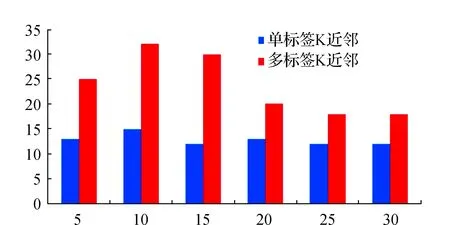
图4 单标签K近邻算法与多标签K近邻算法的比较Fig.4 Comparison of single label and multiple labels K neighbor algorithm
在图4中, 横坐标为k的取值, 纵坐标为在k的取值下所预测出的蛋白质的功能的个数。由图4可见,当k=10, 两种算法预测出的功能个数都是最多的,因此在之后的实验中取值k=10。
用4组评价指标判断k在不同取值下对蛋白质功能预测准确性的影响,见图5。
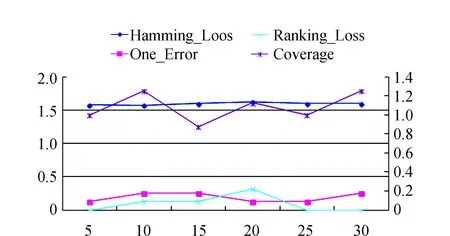
图5 不同k值对算法性能的影响Fig.5 Influence of different k values
在提取注释模式时, 将此方法与传统的方法进行比较, 传统的提取注释模式的方法未考虑转移概率对算法性能的影响, 只统计了与蛋白质相关的已知蛋白质的功能的个数, 对比结果见图6。
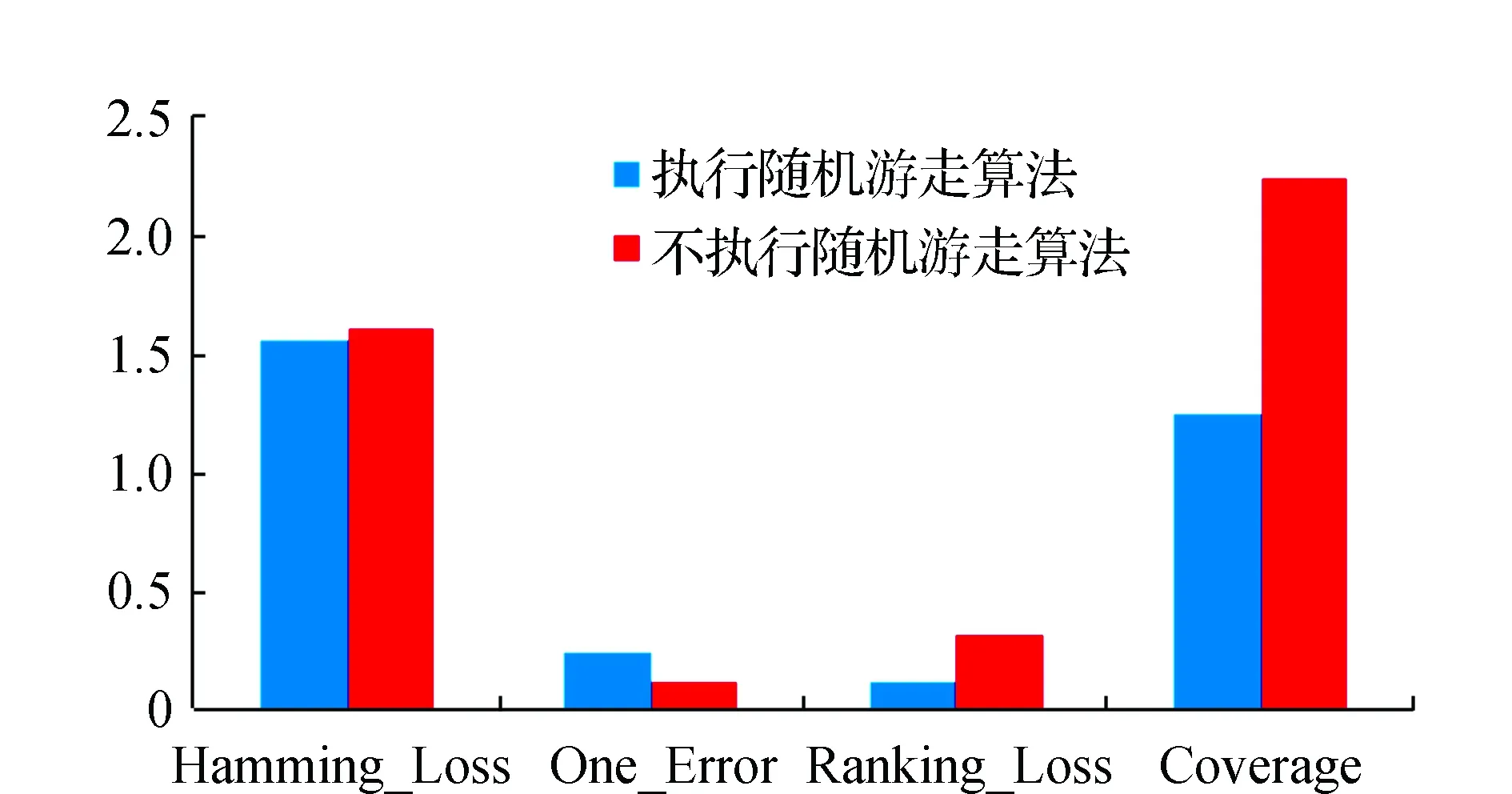
图6 两种提取注释模式方法对比Fig.6 Comparison of two annotation methods
由图6可见,所采用的提取注释模式的方法要优于传统的方法。
在此算法中假设节点度数大的点在网络中相对更重要一些, 也就是说在网络中如果这些相对重要的点为已知功能的蛋白质, 预测结果会更好。选择了同样大小的预测集, 只是在这个预测集中节点的度数都是相对较小的。原来的预测集是把节点的度数较大的点作为未知功能的蛋白质, 用这两组预测集做实验进行对比, 结果见图7。

图7 相同的网络中不同的预测集结果对比Fig.7 Comparison of two different dataset in the same net
由图7可见,换了预测集之后的结果要明显优于之前的结果。
用一型糖尿病构建的网络中实现本文算法, 在这个网络中一共有911个蛋白质, 取出1/7蛋白质做为未知的, 之后用网络中其它的已知功能的蛋白质来预测这些蛋白质的功能。 这1/7蛋白质共有2 960个功能, 用4组评价指标判断k在不同取值下对蛋白质功能预测准确性的影响,结果见图8。
由图8可见,当k为400时各项指标的值综合表现最好。

图8 不同k值对算法性能的影响Fig.8 Influence of different k values
大网络上对多标签的K近邻算法与单标签的算法进行了比较, 见图9。 从标签的K近邻算法所得到的结果要明显优于单标签的K近邻算法。 经过实验验证, 证实本文算法能够有效地预测出蛋白质的功能。

图9 单标签K近邻算法与多标签K近邻算法的比较Fig.9 Comparison of single label and multiple labels K neighbor algorithm
6 结 论
主要实现了对未知功能的蛋白质的功能预测, 在完成这项工作的过程中,整合多个相互作用数据库中的数据, 利用一型糖尿病通路数据, 把相互作用数据库中与一型糖尿病通路数据有互作关系的数据取出来, 实现了随机游走在蛋白质互作网络中的应用, 以互作网络中已知功能的蛋白质为起始点, 计算已知功能的蛋白质与未知功能蛋白质的关联程度, 得到在稳态分布下的概率向量, 即邻居信息。把邻居模式转换成注释模式, 利用已知功能的蛋白质实现未知功能蛋白质的功能的预测。
利用K近邻算法, 把已知功能的蛋白质的注释模式分为训练样本集, 未知功能的蛋白质的注释模式分为待测样本集, 通过把待测样本集中的注释模式与训练样本集中的进行比较, 得到与之最近的k个。 利用多标签分类的KNN算法, 实现蛋白质多标签的功能分类。
[1]Gavin A C, Bösche M, Krause R, et al.Functional organization of the yeast proteome by systematic analysis of protein complexes[J]. Nature, 2002, 415(6 868): 141-147.
[2]Vazquez A, Flammini A, Maritan A, et al.Global protein function prediction from protein-protein interaction networks[J]. Nature biotechnology, 2003, 21(6): 697-700.
[3]Borges H B, Nievola J C.Multi-Label Hierarchical Classification using a Competitive Neural Network for protein function prediction[C].Neural Networks (IJCNN), The 2012 International Joint Conference on,2012: 1-8.
[4] Jiang J Q, Mcquay L J. Predicting protein function by multi-label correlated semi-supervised learning[J]. Computational Biology and Bioinformatics, IEEE/ACM Transactions on, 2012, 9(4): 1 059-1 069.
[6] Chatr-aryamontri A, Breitkreutz B J, Heinicke S, et al. The Biogrid interaction database: 2013 update[J]. Nucleic acids research, 2013, 41(D1): D816-D823.
[7]Benso A, Di Carlo S, Politane G, et al. Combining homolog and motif similarity data with Gene Ontology relationships for protein function prediction[C].Bioinformatics and Biomedicine (BIBM), 2012 IEEE International Conference on. 2012: 1-4.
[8] Lei C, Ruan J. A random walk based approach for improving protein-protein interaction network and protein complex prediction[C]. Bioinformatics and Biomedicine (BIBM), 2012 IEEE International Conference on. 2012: 1-6.
[9] Li P, Heo L, Li M, et al. Protein function prediction using frequent patterns in protein-protein interaction networks[C].Fuzzy Systems and Knowledge Discovery (FSKD), 2011 Eighth International Conference on. 2011, 3: 1 616-1 620.
[10]Vazquez A, Flammini A, Maritan A, et al. Global protein function prediction from protein-protein interaction networks[J]. Nature biotechnology, 2003, 21(6): 697-700.
[11]Yu G, Rangwala H, Domeniconi C, et al.Protein Function Prediction using Multi-label Ensemble Classification[J]. IEEE/ACM Transactions on Computational Biology and Bioinformatics (TCBB), 2013, 10(4): 1-1.
[12]Kamakura N, Takahashi H, Nakamura K, et al. Protein function prediction based on k-cores of interaction networks[C].Bioinformatics and Biomedical Technology (ICBBT), 2010 International Conference on. 2010: 211-215.
Prediction of protein function based on random walk algorithm design and implementation
MA Ji-Quan1, JIA Cui-Cui1,ZHANG Jun-Jie2
(1.School of Computer Science and Technology,Heilongjiang University, Harbin 150080, China;2.Institute of Aerosprace Electronics Technology of Shandong,Yantai 264670,Shandong,China)
In the process of constructing the protein-protein interaction network is starting from the pathway data, data integration using pathway of protein interaction networks.A prediction method for protein function is presented based on random walk, in this algorithm, the proteins of known function as a starting point is provided, for the random walk in the protein interaction network of neighborhood information, annotation schema information is converted, and according to the known function protein function to extract the unknown function protein annotation mode.After, using the traditional KNN algorithm to find the unknown function protein theknearest neighbors from the training set.Last, combined with KNN algorithm for multi label classification, the maximum posteriori probability prediction function label unknown function protein is based.The experiments in which we construct protein interaction network, the results show that the proposed scheme can predict the protein function effectively.
prediction of protein function;protein-protein interaction networks;random walk;protein function annotation;KNN;multi-label classification
10.13524/j.2095-008x.2015.03.050
2015-08-16
黑龙江省自然科学基金资助项目(F201248)
马吉权(1975-),男,黑龙江伊春人,副教授,博士,硕士研究生导师,研究方向:计算机视觉、数字图像处理和生物信息技术,Email: majiquan@hlju.edu.cn。
TP311.13
A
2095-008X(2015)03-0073-06
猜你喜欢
中学生数理化·中考版(2022年6期)2022-06-05 06:49:10
新高考·高一数学(2022年3期)2022-04-28 07:02:46
中学生数理化·中考版(2021年6期)2021-11-22 07:52:30
新世纪智能(数学备考)(2021年4期)2021-08-06 09:04:50
新世纪智能(数学备考)(2021年4期)2021-08-06 09:04:50
中学生数理化(高中版.高考数学)(2021年1期)2021-03-19 08:28:36
车迷(2018年11期)2018-08-30 03:20:32
海峡姐妹(2018年3期)2018-05-09 08:21:02
公民与法治(2016年10期)2016-05-17 04:12:58
高中生学习·高三版(2016年9期)2016-05-14 09:12:05
