
Dysferlinopathy 的免疫病理与基因突变分析
2015-03-11 05:55董明睿舒晓明焦劲松刘尊敬郭淮莲
中风与神经疾病杂志 2015年6期
董明睿,汪 伟,卢 昕,舒晓明,焦劲松,刘尊敬,郭淮莲
肌纤维膜蛋白Dysferlin 缺陷导致肌浆内囊泡运输障碍及膜修复异常产生肢带型肌营养不良2B(limb-girdle muscular dystrophy 2B,LGMD2B)、Miyoshi 肌病(miyoshi myopathy,MM)[1]和以胫前肌发病的远端肌病(distal myopathy with anterior tibial onset,DMAT)[2]等临床表型,称为Dysferlinopathy,以常染色体隐性方式遗传。本病发病机制主要涉及膜损伤后再修复途径的破坏、T 小管的发育与维持、内源性炎症因子的激活以及肌细胞融合和成熟延迟等因素[3,4]。与其他类型肌营养不良相比较,Dysferlinopathy 有较多的炎性细胞浸润,呈现坏死性肌肉病病理特点,易与多发性肌炎、免疫介导性坏死性肌肉病相混肴造成误诊,导致患者接受了无效的皮质类固醇激素治疗。因此对本病患者进行完善的免疫组织化学染色及基因检测,明确其免疫病理特点及确定基因突变点具有重要意义。
1 资料和方法
1.1 病例收集 收集2013 年1 月-2015 年4月中日友好医院神经内科和风湿免疫科因四肢肌肉萎缩无力伴(或)肌酸激酶(CK)升高而就诊的患者,详细采集病史,包括性别、年龄、发病年龄、首发及主要症状、病情进展程度、生长发育史、家族史及除骨骼肌外其他器官受累情况如中枢神经系统、心脏、周围神经等。
1.2 肌肉活检 所有患者签署肌肉活检知情同意书。采取开放式肌肉活检,活检部位:肱二头肌、股四头肌、胫前肌或腓肠肌。新鲜肌肉组织浸入经液氮冷却的异戊烷速冻后在冰冻切片机切片。肌肉组织病理染色包括:苏木素-伊红(H&E)、改良Gomori(MGT)、细胞色素C 氧化酶(COX)、琥珀酸脱氢酶(SDH)、高碘酸Schiff 反应(PAS)、油红“O”(ORO)、还原型辅酶I 四氮唑还原酶(NADH)、三磷酸腺苷酶(ATPase)染色。IHC 染色:一抗包括抗Dysferlin、MHC-1、CD68、CD3、CD20、CD4、CD8、dystrophin-C、-rod、-N、sarcoglycan-alpha、-beta、-gamma、-delta 抗 体。每次IHC 染色将PBS 缓冲液替代一抗作为阴性对照;非肌营养不良、非肌炎肌肉组织作为正常对照。
1.3 PCR-测序法检测DYSF 基因突变 所有患者签署基因检测知情同意书。EDTA 抗凝管留取患者外周血5 ml。采用标准酚-氯仿法提取基因组DNA。从人类基因组数据库Gen Bank 中获得DYSF的序列,应用Primer3 软件分别对基因的第1~55 号外显子设计引物。PCR 反应体系(20 μl)中包括:模板DNA 1 μl、2×Buffer(Mg2+)10 μl、引 物(2.5 mmol)各0.4 μl、dNTPs(2.5 mmol)4 μl、Taq 酶(5 U/μl)0.4 μl、水3.8 μl。反应条件:95 ℃热变性3 min后,95 ℃30 s、60 ℃40 s、68 ℃3 min 30 s 共35个循环,68 ℃延长5 min。使用Multi Screen PCR 96-Well Plate(Millipore)对PCR 产物进行纯化后测序。
2 结果
结合患者临床特点、遗传方式、肌肉组织IHC染色及基因检测结果,共收集4 例Dysferlinopathy 患者。所有患者均符合以下特点:临床以近端、远端或近远端同时受累的肌肉萎缩无力伴/或高CK 血症;遗传方式为常染色体隐性遗传;肌肉活检IHC 染色中dysferlin 蛋白缺乏;基因测序证实含有DYSF 基因复合杂合或纯合突变。
2.1 临床资料 患者1:19 岁男性。患者一年半前感冒后出现全身无力,2 个月后检查发现CK 明显升高,并出现上下楼梯困难,当地医院诊断为“多发性肌炎”并给予皮质类固醇激素治疗。治疗后肢体无力症状及CK 水平均无改善。症状逐渐加重,于我院就诊时走路无力、蹲下站起困难、举物困难。查体:四肢肌力Ⅳ级,无肌肉萎缩,腱反射正常。CK:7000~10854 IU/L。无家族史。肌电图:肌源性损害。患者2:28 岁女性。3 y 前无明显诱因出现上楼梯费力,无力程度进行性加重,近半年自觉小腿细。自幼跑步时足跟不能着地。父母、姐及儿子体健。查体:双上肢肌力正常、无肌萎缩,双侧髂腰肌肌力Ⅴ级、双股四头肌肌力Ⅳ级、双足背屈Ⅴ-级、双足趾屈Ⅳ-级。双小腿肌肉萎缩。双上肢腱反射正常引出、双侧膝跟腱反射未引出。CK:10000~16226 IU/L。无家族史。肌电图:肌源性损害。患者3:15 岁女性。4 y 前开始反复发作双侧大腿乏力,伴双下肢紧绷感。逐渐出现蹲起及上下楼梯困难。查体,双下肢近端肌力Ⅳ级,余肢体肌力正常,无肌萎缩,腱反射正常。CK:4300~7600 IU/L。肌电图:肌源性损害。患者4:19 岁女性。2 y 前体检发现CK 升高(5000 IU/L),不伴肌肉无力,于当地医院接受激素和免疫抑制剂治疗。治疗后CK 水平无明显下降,最高达16000 IU/L。1 y 前出现对称性双下肢无力,上楼梯困难,1 y 来无力程度无明显加重。患者自幼体育成绩较好。无家族史。查体:颈肌及双上肢肌力正常,双下肢肌力Ⅳ-级,无肌萎缩,四肢腱反射对称引出。肌电图:右侧髂腰肌MUP 多相波增多、可见卫星电位,右侧股四头肌、胫前肌MUP 多相波增多。
2.2 一般病理特点及IHC 染色 4 例Dysferlinopathy 患者肌肉活检的共同特点是肌纤维直径变异增加、可见坏死及再生肌纤维、新鲜坏死肌纤维呈均质样改变,符合坏死性肌肉病样病理表现。无或仅伴随轻度肌内衣纤维化,均有肌纤维内脂滴轻度增加。4 例患者的dystrophin-C、-rod、-N 以及sarcoglycan-alpha、-beta、-gamma、-delta 均表达阳性。有3例患者的IHC 染色显示炎细胞以CD68+细胞为主;MHC-1 散在或弥漫表达于细胞膜,包括非坏死及再生肌纤维膜;C5b-9 补体主要沉积在坏死肌纤维内部(见表1、见图1)。未见CD20+细胞。
2.3 DYSF 基因突变检测结果 4 例患者中共发现8 种DYSF 突变:包括移码突变c.2693delG;错义突变c.757C>T、c.5511C>A、c.5516A>T、c.863A>T;剪切突变c.937+1G>A、c.1181-2A>G;无义突变c.1617C>A。其中5 种为新发突变(见表2)。3 种新发错义突变在种属进化中具有保守性(见图2)。

表1 Dysferlinopathy 患者一般病理特点和免疫组化染色

表2 Dysferlinopathy 患者的DYSF 突变
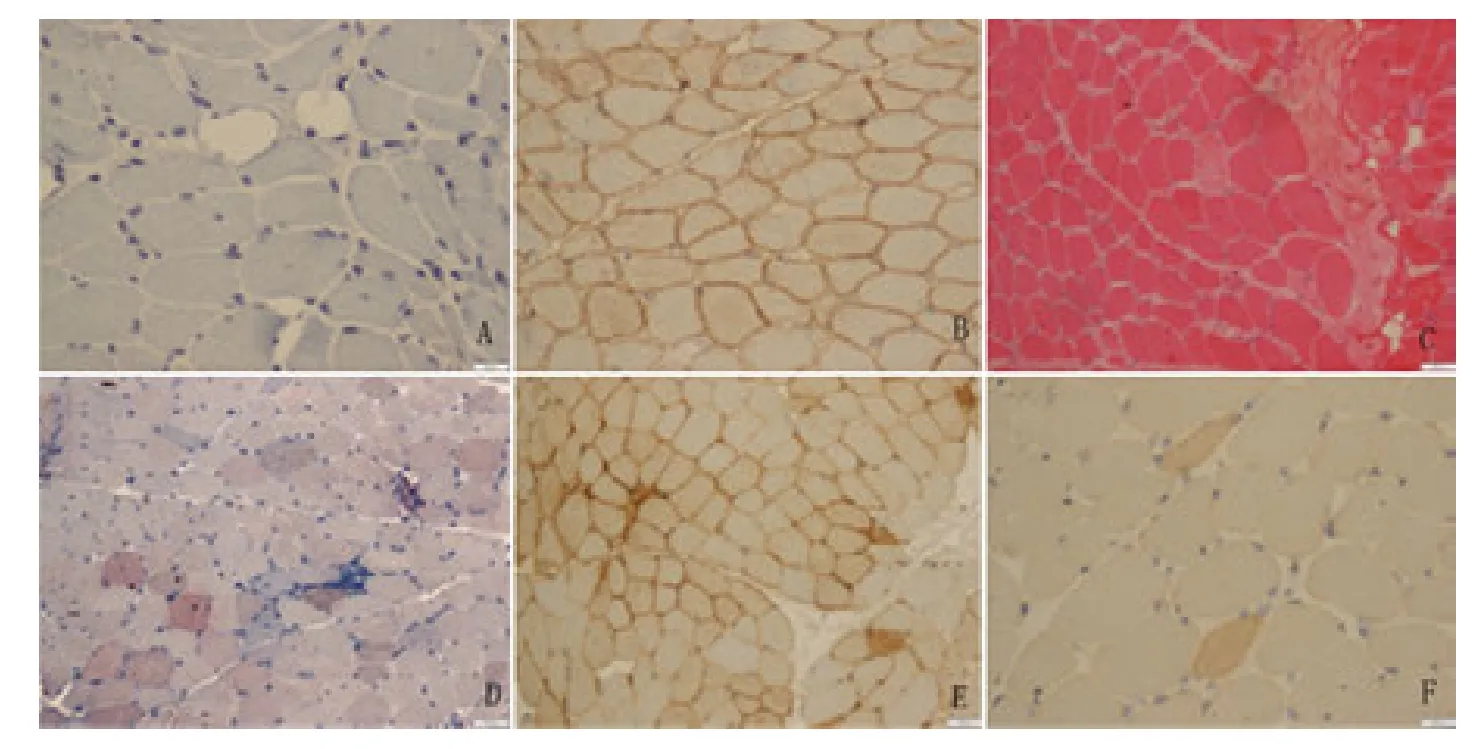
图1 A:患者1 IHC 染色肌纤维膜dysferlin 阴性;B:皮肌炎患者dysferlin 在肌纤维膜阳性表达(做为阳性对照);C:患者1 HE 染色显示少数坏死肌纤维;D:患者4 ORO 染色显示肌纤维内轻度脂滴增多;E:患者4 MHC-1 在多数肌纤维膜阳性表达;F:患者4 C5b-9 补体在坏死肌纤维表达

图2 患者1~3 的测序图
3 讨论
DYSF 位于常染色体2p13 区,由55 个外显子组成,编码分子量230 kDa 的跨膜蛋白dysferlin,后者大量表达在骨骼肌、心肌、脑、胎盘组织[5]以及单核细胞和巨噬细胞[6,7]。Dysferlin 蛋白C-端为跨膜结构域、N-端及其它蛋白结构均位于细胞胞浆内,参与肌肉收缩、以及细胞膜损伤部位囊泡聚集、膜融合过程,与肌细胞膜的再生和修复相关[8]。Dysferlin 参与的浆膜破损后修复过程是通过钙离子触发的、溶酶体参与的各种细胞内囊泡的胞吐作用完成的。经过囊泡-囊泡、囊泡-浆膜的融合过程,大囊泡快速聚集在损伤处,转运“膜补丁”以修补破损的膜结构[3]。Dysferlin 缺乏导致ATP 或其他内源性危险因子如S-100 蛋白、热休克蛋白、HMGB1(High-mobility group box-1)释放,这些因子通过toll-like 受体或P2X7 受体激活了炎性途径[4]。
由于dysferlin 位于细胞膜并与钙蛋白酶-3、小窝蛋白-3 临近并相互作用,因此继发性的dysferlin 蛋白缺乏也见于钙蛋白酶-3 缺陷导致的LGMD2A[9]和小窝蛋白-3 缺陷导致的LGMD1C[10]。因此对通过IHC 染色疑诊的Dysferlinopathy 病例进行基因检测是必要的。在本组4 例患者中,dysferlin 缺失都是由DYSF 基因突变导致的,未发现继发性dysferlin 缺乏。当使用Western blot 方法测量肌肉组织中dysferlin 表达量减少到正常水平的20%以下时,通常是由于DYSF 基因突变所导致,用这个标准诊断Dysferlinopathy 的准确率达到100%[11]。如果未进行肌肉活检,也可以测量血液中单核细胞的dysferlin 表达来代替肌肉组织的检测,这种检测方法使本病的诊断更便捷简单[7]。目前国内大多数实验不应用Western blot 方法进行临床诊断。
Dysferlinopathy 均以隐性方式遗传。我们共发现8 种突变,其中的移码突变c.2693delG、错义突变c.5511C>A(D1837Q)、c.5516A>T(Y1839F)、c.863A>T(D288V)、无义突变c.1617C>A 5 种是首次报道的新发突变。无义突变导致mRNA 翻译提前终止、剪切突变导致外显子跳跃(Exon skipping),因此均被公认具有致病性,3 个新发的错义突变的致病性具有不确定性。通过Polyphen2 和SIFT 功能预测,D1837Q、Y1839F、D288V3 个氨基酸替换均被预测为可能致病和致病;DYSF 第288、1837、1839 位氨基酸序列在种属间具有高度的保守性,以上均是错义突变具有致病性的重要依据。患者2 含有3 种DYSF 突变,除13 号外显子剪切位点处的突变外,另外两处突变为位置临近的两处错义突变,两个错义突变之一可能不具有致病性。
本病多数患者在10~20 岁之间起病,本组患者平均发病年龄17.5 岁。患者2 以远端肌群无力萎缩起病,临床符合MM;患者3 以四肢近端无力为主,临床符合LGMD2B;其他2 例同时出现近、远端肌肉受累。患者2 自幼出现跑步时足跟不能着地的运动发育异常表现,其他3 例在婴幼儿及少年期运动发育正常、体育成绩良好。根据Klinge 等观察:大多数Dysferlinopathy 患者在发病之前肌肉力量正常、体力活动方面表现良好[12],而其他肢带型肌营养不良在明显的肌无力症状出现前常有体育运动成绩欠佳等运动发育异常表现。除具有肌营养不良样病理特点外,Dysferlinopathy 常呈现明显的炎症表现。在3 例患者的IHC 染色,我们观察到MHC-1 在肌纤维膜散在或弥漫表达,包括非坏死肌纤维膜,提示免疫因素参与了疾病的病理过程。本病中炎性细胞以巨噬细胞为主,而免疫介导性坏死性肌肉病以细胞毒性CD4+T 细胞为主,多发性肌炎以CD8+T 淋巴细胞侵入MHC-1 阳性非坏死肌纤维为特点[13]。因此,CD4、CD8、CD68、MHC-1 等IHC 染色有助于Dysferlinopathy 与原发性炎性肌肉病相鉴别。细胞表面的补体C5b、6、7、8、9 形成膜表面的通道结构(MAC 膜攻击复合体),造成膜的穿孔损伤。C5b-9 补体主要位于坏死肌纤维内部[14],这种MAC 的表达可见于大多数肌肉疾病,因此被认为在Dysferlinopathy 发病过程中不起关键作用。膜修复受损也导致脂肪从包裹的膜中释放并在局部沉积,当脂肪细胞在肌纤维聚集,更多的甘油通过脂肪细胞产生,加速了肌病的病理生理过程并最终导致三联体失去功能、改变钙离子稳态,进一步加重肌肉无力[15]。本组中4 例患者肌肉组织均有轻度的脂滴增多,这个结果从一定程度上支持Dysferlinopathy 中继发于膜修复损伤的脂肪转运障碍参与病理生理过程的理论。这种脂滴增多的情况在其他的肌营养不良出现并不广泛。
很多研究者基于对Dysferlinopathy 发病机制的认知,在动物模型中尝试了基因治疗、细胞治疗、小分子药物治疗[16]以及针对炎症反应的皮质类固醇激素、免疫抑制及IvIg 治疗[17]。尽管到目前为止临床上仍无有效的治疗方法,但正确的诊断可以使患者避免因误诊而接受错误的治疗。这4 例患者中的3 例最初被诊断为“肌炎”并接受了皮质类固醇激素治疗。详尽的病史、完善的肌肉组织学及IHC 染色和基因检测对肌肉病诊断具有重要意义。
[1]Liu J,Aoki M,Illa I,et al.Dysferlin,a novel skeletal muscle gene,is mutated in Miyoshi myopathy and limb girdle muscular dystrophy[J].Nat Genet,1998,20:31-36.
[2]Illa I,Serrano-Munuera C,Gallardo E,et al.Distal anterior compartment myopathy:a dysferlin mutation causing a new muscular dystrophy phenotype[J].Ann Neurol,2001,49:130-134.
[3]McDade JR,Michele DE.Membrane damage-induced vesicle-vesicle fusion of dysferlincontaining vesicles in muscle cells requires microtubules and kinesin[J].Hum Mol Genet,2013,23:1677-1686.
[4]Rawat R,Cohen TV,Ampong B,et al.Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle[J].Am J Pathol,2010,176:2891-2900.
[5]Anderson LV,Davison K,Moss JA,et al.Dysferlin is a plasma membrane protein and is expressed early in human development[J].Hum Mol Genet,1999,8:855-861.
[6]De Luna N,Freixas A,Gallano P,et al.Dysferlin expression in monocytes:a source of mRNA for mutation analysis[J].Neuromuscul Disord,2007,17:69-76.
[7]Ho M,Gallardo E,McKenna-Yasek D,et al.A novel,blood-based diagnostic assay for limb girdle muscular dystrophy 2B and Miyoshi myopathy[J].Ann Neurol,2002,51:129-133.
[8]Bansal D,Miyake K,Vogel SS,et al.Defective membrane repair in dysferlin-deficient muscular dystrophy[J].Nature,2003,423:168-172.
[9]Anderson LV,Harrison RM,Pogue R,et al.Secondary reduction in calpain-3 expression in patients with limb girdle muscular dystrophy type 2B and Miyoshi myopathy(primary dysferlinopathies)[J].Neuromuscul Disord,2000,10:553-559.
[10]Matsuda C,Hayashi YK,Ogawa M,et al.The sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal muscle[J].Hum Mol Genet,2001,10:1761-1766.
[11]Cacciottolo M,Numitone G,Aurino S,et al.Muscular dystrophy with marked Dysferlin deficiency is consistently caused by primary dysferlin gene mutations[J].Eur J Hum Genet,2011,19(9):974-980.
[12]Klinge L,Aboumousa A,Eagle M,et al.New aspects on patients affected by dysferlin deficient muscular dystrophy[J].J Neurol Neurosurg Psychiatry,2010,81:946-953.
[13]Gallardo E,Rojas-Garcia R,de Luna N,et al.Inflammation in dysferlin myopathy:immunohistochemical characterization of 13 patients[J].Neurology,2001,57:2136-2138.
[14]Confalonieri P,Oliva L,Andreetta F,et al.Muscle inflammation and MHC class up-regulation in muscular dystrophy with lack of dysferlin:an immunopathological study[J].J Neuroimmunol,2003,142(1~2):130-136.
[15]Demonbreun AR,Rossi AE,Alvarez MG,et al.Dysferlin and myoferlin regulate transverse tubule formation and glycerol sensitivity[J].Am J Pathol,2014,184(1):248-259.
[16]Kobayashi K,Izawa T,Kuwamura M,et al.Dysferlin and animal models for dysferlinopathy[J].J Toxicol Pathol,2012,25(2):135-147.
[17]Angelini C,Peterle E,Gaiani A,et al.Dysferlinopathy course and sportive activity:clues for possible treatment[J].Acta Myol,2011,30(2):127-132.
猜你喜欢
广西医科大学学报(2022年5期)2022-06-07
昆明医科大学学报(2022年3期)2022-04-19
肉类研究(2020年9期)2020-12-14
科技资讯(2020年3期)2020-04-07
中南医学科学杂志(2019年6期)2019-12-05
中国循证儿科杂志(2017年6期)2018-01-23
肉类研究(2017年8期)2017-11-16
汽车维护与修理(2016年3期)2016-02-28
汽车维护与修理(2015年6期)2015-02-28
汽车维护与修理(2015年5期)2015-02-28
