
G93A突变的hSOD1基因对Nrf2/ARE信号通路的影响
2015-03-11 08:26:20亓法英王富敏于继徐车峰远赵孔波
中国临床医学 2015年1期
亓法英 王富敏 于继徐 车峰远 赵孔波
(山东省临沂市人民医院神经内科,*急救中心,山东临沂 276003)
·论著·
G93A突变的hSOD1基因对Nrf2/ARE信号通路的影响
亓法英王富敏于继徐车峰远赵孔波*
(山东省临沂市人民医院神经内科,*急救中心,山东临沂276003)
摘要目的: 探讨G93A突变的hSOD1 (human Cu/Zn superoxide dismutase) 基因在肌萎缩侧索硬化转基因细胞模型NSC-34细胞中对Nrf2/ARE信号通路的影响。方法:将构建好的质粒hSOD1-pcDNA3.1(-)、hSOD1-G93A-pcDNA3.1(-)和pcDNA3.1(-)转染至肌萎缩侧索硬化转基因细胞模型NSC-34细胞内,根据转染质粒的不同分为4组:正常组、空转组、野生组和突变组。通过检测细胞内脂质过氧化产物丙二醛(malondialdehyde, MDA)的含量来判断细胞氧化损伤的程度;检测线粒体膜通透性;通过Western blotting检测细胞中Nrf2、抗氧化酶的蛋白表达水平,以反映Nrf2/ARE信号通路在各组的活化程度。结果:转染hSOD1-G93A-pcDNA3.1(-)的NSC-34细胞(突变组)内氧化应激水平增高,同时线粒体通透性增加,提示线粒体受损伤;且Nrf2以及Nrf2/ARE信号通路下游效应分子血红素氧合酶-1(heme oxygenase-1, HO-1)和NAD(P)H醌氧化还原酶-1[NAD(P)H: quinone oxidoreductase 1, NQO1]的蛋白表达水平均降低(P<0.05)。突变组细胞胞浆中Nrf2表达明显减少(P<0.05),而突变组和野生组细胞核中Nrf2增多,突变组细胞更为显著(P<0.05)。结论:hSOD1基因的G93A突变使细胞Nrf2/ARE信号通路受损,降低了细胞的抗氧化能力,加重了线粒体损伤。
关键词肌萎缩侧索硬化;NSC-34细胞;Nrf2/ARE通路
肌萎缩侧索硬化(amyotrophic lateral sclerosis, ALS)是一种选择性侵犯上、下运动神经元的慢性致死性神经系统变性疾病,临床主要表现为进行性发展的肌肉无力、萎缩、肌束震颤、腱反射亢进和病理征阳性。ALS患者中,90%~95%为散发病例,5%~10%的患者有家族史。目前,ALS的发病机制不明,缺乏有效的治疗方法,患者多于发病3~5年后死于呼吸肌麻痹[1-2]。
目前认为,ALS患者的运动神经元进行性丢失是由一系列复杂的相互作用机制所致。这些机制包括氧化应激、谷氨酸兴奋性毒性、线粒体功能障碍、细胞骨架异常、蛋白聚集以及遗传因素等,其中,氧化应激是其主要因素[3]。有研究[4-5]在约20%的家族性ALS患者中发现了铜锌超氧化物歧化酶(Cu/Zn SOD)的突变;转染突变的人类SOD1基因的小鼠可以表现出ALS样症状。然而,迄今为止,对于病因已经明确的转染突变的人类SOD1基因的动物毒性致病机制尚不清楚。近年研究发现,核因子E2相关因子2(nuclear factor erythroid 2-related factor 2, Nrf2)通过与抗氧化反应元件(antioxidant response element, ARE)的相互作用来调节编码抗氧化酶和抗氧化蛋白(包括醌氧化还原酶1和血红素氧合酶),这是迄今为止发现的最重要的内源性抗氧化应激通路。有研究[6-7]发现,在不同的组织和器官中,Nrf2/ARE信号通路的激活能够保护机体或者减少细胞损伤。但是,关于Nrf2/ARE信号通路在ALS运动神经元变性的发病过程中发挥的作用目前仍未完全阐明。
NSC-34细胞模型在国外已经被广泛应用于ALS的研究[8-9]。本研究应用NSC-34细胞构建的ALS细胞模型,观察G93A突变的人SOD1基因(human SOD1-G93A, hSOD1-G93A)对NSC-34细胞内源性抗氧化通路Nrf2/ARE信号通路的影响,以探讨ALS的发病机制,为ALS治疗药物的筛选和治疗提供更多的依据。
1资料与方法
1.1实验材料
NSC-34细胞系由河北医科大学第二医院神经病学实验室惠赠;分别稳定转染hSOD1-pcDNA3.1(-)、hSOD1-G93A-pcDNA3.1(-)和pcDNA3.1(-)质粒的NSC-34细胞系为本实验室前期构建;NAD(P)H醌氧化还原酶-1[NAD(P)H: quinone oxidoreductase 1, NQO1]多克隆抗体、血红素氧合酶-1(heme oxygenase-1, HO-1)多克隆抗体、核转录因子E2相关因子2(nuclear factor E2-related factor 2, Nrf2)多克隆抗体均购自美国Santa Cruz公司;总蛋白提取试剂盒、胞浆/胞核蛋白提取试剂盒购自江苏省南京凯基生物科技发展有限公司。
1.2方法
1.2.1细胞培养实验共分为4组:正常NSC-34细胞(正常组)、转染pcDNA3.1(-)质粒的NSC-34细胞(空转组);转染hSOD1-pcDNA3.1(-)质粒的NSC-34细胞(野生组);转染hSOD1-G93A-pcDNA3.1(-)质粒的NSC-34细胞(突变组),均为本实验室前期构建。进行细胞培养时,将4种细胞系从液氮中取出,置于37℃水浴箱中迅速融化,然后转移至25 cm2玻璃培养瓶中,加入5 mL含10%胎牛血清的DMEM完全培养基并混匀,将培养瓶盖旋至半松状态,置于37℃、CO2体积分数为5%的培养箱内培养;接种4~6 h后更换培养液,之后每 2~3 更换一次培养液。NSC-34细胞的免疫组化(anti-SIM2抗体,ab94630,英国Abcam公司)图见图1。
图1NSC-34细胞的免疫组化图片(anti-SIM2染色)
1.2.2细胞丙二醛检测通过检测脂质过氧化产物丙二醛(malondialdehyde, MDA)的含量来反应细胞脂质过氧化的水平。在过氧化脂质降解产物中的MDA可与硫代巴比妥酸缩合,形成红色产物,在532 nm波长处有最大吸收峰。根据MDA检测试剂盒的说明书操作,在室温下将各组细胞破碎并与反应液混合,于95℃水浴箱内水浴40 min,然后迅速用流水冷却。加入96孔板内,测定其在523 nm处的吸光度。
1.2.3蛋白质的提取及浓度测定当细胞融合度达80%~90%时,用预冷的磷酸盐缓冲液(PBS)洗2次,0.25%胰酶消化,1000 r/min、4℃离心10 min。细胞总蛋白的提取按照总蛋白提取试剂盒说明书进行。细胞浆和细胞核蛋白的提取按照胞浆/胞核蛋白提取试剂盒说明书进行。用Bradford法测定蛋白浓度。
1.2.4Western blotting检测蛋白水平取60 μg蛋白,用12%的十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)胶、80 V电泳约30 min,当溴酚蓝过了积层胶后,改用100 V电泳至溴酚蓝达底部。电转移至聚偏二氟乙烯膜(polyvinylidene fluoride, PVDF)膜上,参数为300 mA、2 h。经5%脱脂奶粉封闭1 h后,加一抗:鼠来源单克隆β-actin抗体(1∶500)、兔来源多克隆Nrf2抗体(1∶200)、兔来源多克隆HO-1抗体(1∶200)、羊来源多克隆NQO1抗体(1∶200),4℃过夜。次日,用TBST(Tris buffered saline with Tween-20, PH 7.4)洗涤后加相应的荧光二抗(β-actin二抗为1∶10000,其余均为1∶3000),室温孵育1 h, TBST(PH 7.4)洗涤,加ECL发光液后压片、曝光。最后用成像仪扫描目的条带,计算目的蛋白荧光条带密度与相应β-actin荧光条带密度的比值。
1.2.5线粒体通透性的检测采用线粒体膜通透性转运孔检测试剂盒mitoprobeTMtransition pore assay kit (美国invitrogen公司)。将1 mmol/L的calcein-AM储存液稀释为浓度为2 μmol/L的工作液,即按照1∶500的比例用HBSS/Ca2+溶液稀释。设置阴性对照组、线粒体摄入calcein-AM的对照组以及背景强阳性对照组。向6孔板培养细胞的培养液内加入2 μmol/L的calcein工作液5 μL,背景强阳性对照组内加入离子霉素(ionomysin)和CoCl2各 5 μL,其余各个组内再加入CoCl25 μL(包括线粒体对照管),37℃孵育15 min,加入HBSS/Ca2+溶液清洗,1 h内完成荧光定量分析。
1.3统计学处理
2结果
2.1hSOD1-G93A-pcDNA3.1(-)过表达可以显著增加细胞内氧化应激水平统计学分析显示,转染hSOD1-G93A-pcDNA3.1(-)质粒的NSC-34细胞(突变组)中MDA的含量明显高于其他3组细胞(P<0.05);而转染pcDNA3.1(-)的NSC-34细胞(空转组)和转染hSOD1-pcDNA3.1(-)质粒的NSC-34细胞(野生组)与正常的NSC-34细胞(正常组)相比,细胞内MDA的含量没有明显差异。见表1。
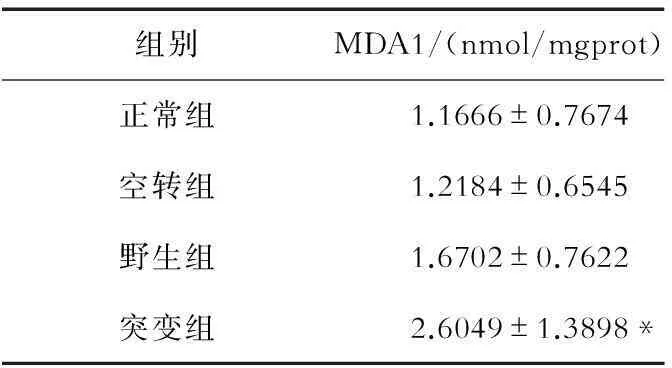
表1 4组细胞内MDA的含量
注:与正常组、空转组和野生组比较,*P<0.05
2.2hSOD1-G93A-pcDNA3.1(-)过表达可以明显增加细胞浆内线粒体膜的通透性在转染hSOD1-G93A-pcDNA3.1(-)质粒的NSC-34细胞(突变组)中,细胞浆线粒体内calcein荧光强度较正常组、空转组及野生组显著下降,提示突变组中线粒体通透性增加(P<0.05)。野生组与空转组的NSC-34细胞线粒体内Calcein荧光强度与正常组NSC-34细胞相比,均无统计学差异(P>0.05),提示野生组及空转组中没有明显的线粒体通透性改变。见图2。
2.3hSOD1-G93A-pcDNA3.1(-)过表达可以下调Nrf2以及HO-1、NQO1蛋白的表达水平在转染hSOD1-G93A-pcDNA3.1(-)质粒的 NSC-34 细胞中,细胞内总Nrf2以及HO-1、NQO1蛋白的表达均低于正常组(P<0.05)。转染pcDNA3.1(-)质粒和转染hSOD1-pcDNA3.1(-)质粒的NSC-34细胞与正常NSC-34细胞相比,Nrf2及HO-1、NQO1蛋白的表达无明显变化(P>0.05)。见图3。
2.4hSOD1-G93A-pcDNA3.1(-)过表达可以下调胞浆Nrf2的表达水平并增加Nrf2蛋白的核转位突变组细胞胞浆中Nrf2蛋白的表达与其他3组细胞相比明显减少(P<0.05),见图3,而其他3组细胞胞浆中Nrf2的表达差异无统计学意义。野生组和突变组细胞胞核中Nrf2蛋白与其他两组相比明显增加,且突变组细胞更为显著(P<0.05),见图4。
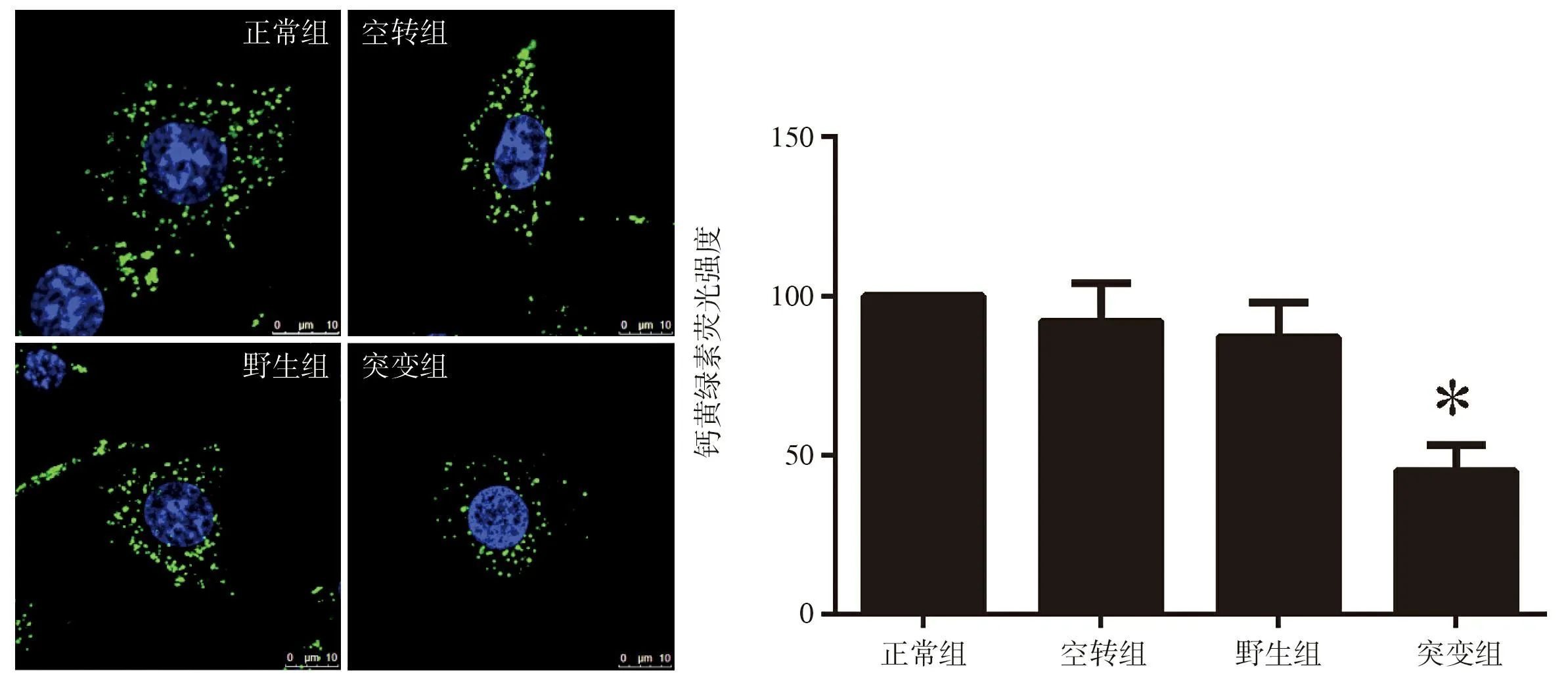
图2hSOD1-G93A对线粒体膜通透性的影响
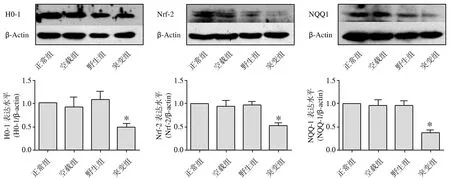
图3 hSOD1-G93A对Nrf2、HO-1、NQO1蛋白表达的影响
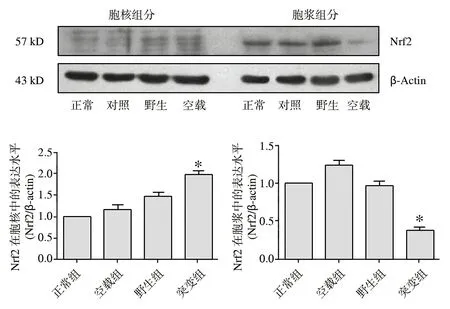
图4 4组细胞的细胞核与细胞浆内Nrf2蛋白水平的变化
3讨论
家族性ALS患者中约20%有SOD1基因突变,把hSOD1-G93A转染入小鼠中,可以使小鼠出现ALS神经系统样症状。为了研究hSOD1-G93A基因的致病机制,我们应用NSC-34细胞过表达hSOD1-G93A基因,观察hSOD1-G93A基因过表达对内源性抗氧化应激通路的影响,结果发现,hSOD1-G93A过表达可以显著增加MDA的水平,提示hSOD1-G93A过表达可以抑制NSC-34细胞的抗氧化和解毒能力。同时,hSOD1-G93A过表达可以使线粒体通透性显著增加。已有研究[10]表明,线粒体通透性的增加与细胞凋亡的发生有关,线粒体膜通透性的增加可以使线粒体内膜中的凋亡诱导因子(apoptosis induce factor,AIF)、caspase-3等释放入胞浆并诱导细胞凋亡。本研究发现,在转染hSOD1-G93A-pcDNA3.1(-)的NSC-34细胞内,抗氧化应激的中枢调节蛋白Nrf2的表达水平明显低于正常对照组,与之相对应,Nrf2-ARE信号通路下游效应因子HO-1和NQO1的蛋白表达水平也明显低于正常对照组。在正常状态下,Nrf2在细胞浆中与其抑制性蛋白Keapl(Kelch-like ECH-associated protein-1)相结合,处于非活性状态。当细胞发生氧化应激时,在氧自由基等的作用下,Nrf2与Keapl蛋白分离,Nrf2由胞浆转位到胞核内,通过与核内ARE顺式作用元件相结合来上调Ⅱ相解毒酶及抗氧化蛋白(包括HO-1和NQO1)的表达[11-13]。HO-1被认为在内源性抗氧化防御过程中起重要作用,因为在HO-1基因敲除鼠模型中,细胞更易受到氧化应激的损伤[14]。NQO1是一种电子还原酶,它能够催化醌类及其衍生物发生还原反应,且反应过程中不会产生单电子还原产物半醌及自由基等氧化产物,这有益于抵御机体代谢引起的氧化应激反应。研究[15]表明,破坏Nrf2分子及其下游信号通路会加重细胞内的氧化损伤、炎性反应和线粒体功能障碍。Ⅱ相酶诱导剂可以通过Nrf2/ARE通路上调HO-1的表达,保护神经元免受氧化应激的损伤。以上证据表明,Nrf2/ARE信号通路可以对抗氧化应激,保护神经元。但是,在突变组NSC-34细胞中Nrf2/ARE信号通路受损,Nrf2、HO-1和NQO1的表达下降,因此,突变组NSC-34细胞中氧化应激水平升高,导致细胞损伤。
为了进一步了解Nrf2/ARE信号通路的改变,我们分别检测了NSC-34细胞浆和细胞核中Nrf2蛋白表达的变化,结果发现,突变组细胞胞浆中Nrf2的表达与其他3组细胞相比明显减少,而正常组、空转组和野生组细胞胞浆内Nrf2的表达无明显差异;野生组和突变组细胞核中Nrf2蛋白与其他两组相比增加,而且突变组细胞增加更显著(P<0.05)。以上结果与Mimoto等[16]在ALS动物模型的运动神经元细胞核中发现的Nrf2积聚是一致的。有研究[17-18]在突变的TDP-43转基因细胞模型中也发现Nrf2核转移,但是其下游抗氧化酶HO-1表达却降低。也就是说,当细胞受到氧化应激时,突变组细胞中有更多的Nrf2由胞浆转移至胞核,但是其下游的抗氧化酶的蛋白表达水平却是降低的,这也说明基因突变导致Nrf2/ARE信号通路受阻,使得下游效应因子的表达不能相应地增加。
综上所述,在ALS模型中,hSOD1-G93A基因使细胞Nrf2/ARE信号通路受损,降低了细胞的抗氧化能力。因此,Nrf2/ARE信号通路有潜力成为治疗ALS的新靶点。
参考文献
[1]Mitchell JD, Callagher P, Gardham J, et al. Timelines in the diagnostic evaluation of people with suspected amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND)-a 20-year review: can we do better?[J]. Amyotroph Lateral Scler, 2010,11(6):537-541.
[2]Kingwell K. Amyotrophic lateral sclerosis: Early involvement of grey matter oligodendrocytes in amyotrophic lateral sclerosis[J]. Nat Rev Neurol, 2013,9(5):238.
[3]Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis[J]. Lancet, 2011,377(9769):942-955.
[4]Riva N, Chaabane L, Peviani M, et al. Defining peripheral nervous system dysfunction in the SOD-1G93A transgenic rat model of amyotrophic lateral sclerosis[J]. J Neuropathol Exp Neurol,2014,73(7):658-670.
[5]Poulletier DGF, Ruffie G, Taxile M, et al. Amyotrophic lateral sclerosis (ALS) and extremely-low frequency (ELF) magnetic fields: a study in the SOD-1 transgenic mouse model[J]. Amyotroph Lateral Scler, 2009,10(5-6):370-373.
[6]Vargas MR, Johnson JA. The Nrf2-ARE cytoprotective pathway in astrocytes[J]. Expert Rev Mol Med,2009,11:e17.
[7]Jian Z, Li K, Song P, et al. Impaired activation of the Nrf2-ARE signaling pathway undermines H2O2-induced oxidative stress response: a possible mechanism for melanocyte degeneration in vitiligo[J]. J Invest Dermatol, 2014,134(8):2221-2230.
[8]Rizzardini M, Mangolini A, Lupi M, et al. Low levels of ALS-linked Cu/Zn superoxide dismutase increase the production of reactive oxygen species and cause mitochondrial damage and death in motor neuron-like cells[J]. J Neurol Sci, 2005,232(1-2):95-103.
[9]Gomes C, Keller S, Altevogt P, et al. Evidence for secretion of Cu, Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis[J]. Neurosci Lett, 2007,428(1):43-46.
[10]Smith MA, Schnellmann RG. Calpains, mitochondria, and apoptosis[J]. Cardiovasc Res, 2012,96(1):32-37.
[11]Neymotin A, Calingasan NY, Wille E, et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis[J]. Free Radic Biol Med, 2011,51(1):88-96.
[12]Ji L, Wei Y, Jiang T, et al. Correlation of Nrf2, NQO1, MRP1, cmyc and p53 in colorectal cancer and their relationships to clinicopathologic features and survival[J]. Int J Clin Exp Pathol, 2014,7(3):1124-1131.
[13]Li L, Dong H, Song E, et al. Nrf2/ARE pathway activation, HO-1 and NQO1 induction by polychlorinated biphenyl quinone is associated with reactive oxygen species and PI3K/AKT signaling[J]. Chem Biol Interact, 2014,209:56-67.
[14]Yu JH, Cho SO, Lim JW, et al. Ataxia telangiectasia mutated inhibits oxidative stress-induced apoptosis by regulating heme oxygenase-1 expression[J]. Int J Biochem Cell Biol, 2015,60C:147-156.
[15]Slocum SL, Kensler TW. Nrf2: control of sensitivity to carcinogens[J]. Arch Toxicol, 2011,85(4):273-284.
[16]Mimoto T, Miyazaki K, Morimoto N, et al. Impaired antioxydative Keap1/Nrf2 system and the downstream stress protein responses in the motor neuron of ALS model mice[J]. Brain Res, 2012,1446:109-118.
[17]Duan W, Guo Y, Xiao J, et al. Neuroprotection by monocarbonyl dimethoxycurcumin C: ameliorating the toxicity of mutant TDP-43 via HO-1[J]. Mol Neurobiol, 2014,49(1):368-379.
[18]Guo Y, Wang Q, Zhang K, et al. HO-1 induction in motor cortex and intestinal dysfunction in TDP-43 A315T transgenic mice[J]. Brain Res,2012,1460:88-95.
基本项目:国家重点临床专科建设项目(编号:财社[2010]305号)
Influences of hSOD1 Gene with G93A Mutation on Nrf2/ARE Signaling Pathway
QIFayingWANGFuminYUJixuCHEFengyuanZHAOKongbo*DepartmentofNeurology,*EmergencyCenter,LinyiPeople′sHospital,Linyi276003,China
AbstractObjective: To investigate the effect of human Cu/Zn superoxide dismutase (hSOD1) gene with G93A mutation on Nrf2/ARE signaling pathway in NSC-34 cell,the transgenic cell model of amyotrophic lateral sclerosis (ALS). Methods: The established plasmids, hSOD1-pcDNA3.1(-), hSOD1-G93A-pcDNA3.1(-), and pcDNA3.1(-) were transfected into NSC-34 cells, the transgenic cell model of amyotrophic lateral sclerosis. The models were divided into four groups according to different transfected plasmids. They were normal group, empty group, wild group and mutation group. The oxidative-stress injury was evaluated by detecting the content of intracellular malondialdehyde(MDA),a lipid peroxidation product. The permeability of mitochondrial membrane was detected. Western blotting was used to determine the intracellular protein expression level of Nrf2 and antioxidase, so as to reveal the activation level of the Nrf2/-ARE signaling pathway in each group. Results: The level of oxidative stress and the mitochondrial permeability increased in the NSC-34 cells transfected with human hSOD1-G93A-pcDNA3.1(-) gene(mutation group,P<0.05), which implied impairment of mitochondrias. The protein expression level of Nrf2, significantly decreased in NSC-34 cells transfected with hSOD1-G93A gene(P<0.05). So were heme oxygenase-1 (HO-1) and NAD(P)H: quinone oxidoreductase 1 (NQO1), downstream effector molecules of Nrf2-ARE signaling pathway. The expression of Nrf 2 in cytoplasm significantly decreased in mutation group, while Nrf2 expression in cell nucleus significantly increased (P<0.05) in mutation group and wild group, especially in mutation group (P<0.05). Conclusions: The G93A mutation of hSOD1 gene impairs Nrf2/ARE signaling pathway in ALS cell models, reduces the antioxidant ability of cells, and increase the impairment of mitochondrias.
Key WordsAmyotrophic lateral sclerosis; NSC-34 cells;Nrf2/ARE signaling pathway
通讯作者张平安,E-mail:zhangpingan@aliyun.com
中图分类号R34
文献标识码A
猜你喜欢
海洋通报(2021年1期)2021-07-23 01:55:14
生物学通报(2021年4期)2021-03-16 05:41:26
广东医科大学学报(2020年6期)2020-02-06 06:00:38
癌变·畸变·突变(2016年5期)2016-08-22 05:55:18
中华老年多器官疾病杂志(2016年8期)2016-05-14 07:16:56
中国男科学杂志(2016年9期)2016-03-20 15:00:13
结核与肺部疾病杂志(2015年1期)2015-07-18 11:09:22
畜牧兽医学报(2015年3期)2015-07-05 08:22:42
现代检验医学杂志(2015年5期)2015-02-06 01:42:36
癌变·畸变·突变(2014年1期)2014-03-01 04:39:36
