
塞克硝唑的质量标准研究
2015-03-09 10:37何鸽飞郑金凤邱细敏湖南省肿瘤医院长沙1001长沙市第一医院长沙10005湖南省食品药品检验院长沙10001湖南师范大学医学院药学系长沙10005
中国药房 2015年9期
潘 勇,何鸽飞,郑金凤,童 达,邱细敏(1.湖南省肿瘤医院,长沙 1001;.长沙市第一医院,长沙 10005;.湖南省食品药品检验院,长沙 10001;.湖南师范大学医学院药学系,长沙 10005)
塞克硝唑(Secnidazole)化学名是1-(2-羟基丙基)-2-甲基-5-硝基咪唑,具有抗多种厌氧菌和原虫的作用,特别是溶解宿主组织内阿米巴病、贾第鞭毛虫等[1-2]。该药由罗纳普朗克乐安(Rhone-Poulenc Rorer)公司研制,于1980年在瑞士上市。我国塞克硝唑的现有质量标准为国家食品药品监督管理局标准(试行)YBH09922005和国家食品药品监督管理局标准YBH00562010。目前,《美国药典》(USP)33版、《英国药典》(BP)2009版、《日本药典》(JP)15版以及2010年版《中国药典》均未收载该品种。本研究考察了塞克硝唑原料药的性状、鉴别、检查、含量测定等项目,以为2015年版《中国药典》塞克硝唑质量标准的拟订提供参考。
1 材料
1.1 仪器
YRT-3型熔点测定仪(天津天光光学仪器有限公司);UV-2550紫外分光光度计(日本岛津公司);pHS-3C型精密pH计(上海精科仪器有限公司);1100高效液相色谱(HPLC)仪,配有VWD紫外检测器(美国安捷伦公司);GC-2010气相色谱(GC)仪(日本岛津公司);Precisa-120A电子天平(上海分析仪器厂)。
1.2 药品与试剂
塞克硝唑(A公司,生产批号:200910F01、201006F01、201007F14;B公司,生产批号:20071226、20100724、20100725);甲醇、乙腈、二氯甲烷、乙酸乙酯、甲苯为色谱纯,其余试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 性状
根据化学原料药质量标准制定指导原则,性状应分别记述药品的外观、嗅、味、溶解度以及有关物理常数。本研究参照现行的质量标准对塞克硝唑的外观、溶解性、熔点及吸收系数进行了考察和统一。
2.1.1 外观 取本品适量,目视法观察其外观性状。结合6批样品的实际外观,将其外观性状项统一修订为:本品为类白色或微黄色结晶或结晶性粉末,无臭,味苦。
2.1.2 溶解性 取本品适量,依2010年版《中国药典》(二部)凡例试验,结合6批样品的实测结果,将其溶解性行为统一修订为:在甲醇、乙醇、三氯甲烷、丙酮中易溶,在0.1 mol/L盐酸溶液中溶解,在乙醚中略溶,在水中微溶。
2.1.3 熔点 取本品适量,依2010年版《中国药典》(二部)附录ⅥC测定,结合6批样品的实测结果,将本品熔点统一修订为:73~78 ℃。
2.1.4 吸收系数 取本品适量,精密称定,加0.1 mol/L盐酸溶液溶解并稀释制成每1 ml中含塞克硝唑13 μg的溶液,依2010年版《中国药典》(二部)附录ⅣA紫外-可见分光光度法,在277 nm波长处测定吸光度,计算吸收系数。结合6批样品的实际测定值,同时参照国家食品药品监督管理局标准YBH00562010,将吸收系数修订为:331~349。
2.2 鉴别
我国现行的两个质量标准中均收载了化学反应、紫外光谱鉴别和红外光谱鉴别。笔者通过对6批样品进行考察,化学反应和红外光谱鉴别未作修订;紫外光谱鉴别考虑到样品的溶解性,参照国家食品药品监督管理局标准YBH00562010进行了统一:取“2.1.4”项下的溶液,依2010年版《中国药典》(二部)附录ⅣA紫外-可见分光光度法测定,在277 nm波长处有最大吸收,在241 nm波长处有最小吸收。
2.3 检查
根据《化学原料药质量标准制定指导原则》,检查项目的设置应考虑药品的有效性、纯度要求和安全性。笔者参照现行质量标准设置了乙醇溶液的澄清度与颜色、酸碱度、一般杂质检查(硫酸盐、炽灼残渣、重金属)、水分或干燥失重、有关物质及残留溶剂检查项。
2.3.1 乙醇溶液的澄清度与颜色 取本品适量,加乙醇溶解并稀释制成每1 ml中约含20 mg塞克硝唑的溶液。结果,6批样品的溶液均澄清,其所显溶液的颜色均浅于黄色或黄绿色2号标准比色液。
2.3.2 酸碱度 取本品约0.1 g,加水10 ml使溶解,依2010年版《中国药典》(二部)附录ⅥH测定。结合6批样品的实际测定值,同时参照国家食品药品监督管理局标准(试行)YBH09922005,将其酸碱度确定为:pH为5.5~7.5。
2.3.3 硫酸盐 取本品1.0 g,加水100 ml使溶解,滤过,取续滤液40 ml,依2010年版《中国药典》(二部)附录ⅧB检查,与标准硫酸钾溶液2.0 ml制成的对照液(0.05%)比较。结果,6批样品的供试品溶液所显浑浊均浅于对照溶液。
2.3.4 炽灼残渣 取本品1.0 g,依2010年版《中国药典》(二部)附录ⅧN法检查,遗留残渣不得超过0.1%。结果,6批样品均符合规定。
北京市快递运输主要集中在街道、小区及各个单位。调查显示,学生对学校快递业务员的服务相对满意,而小区、街道需要提高。反映的问题主要是送货时间不准确、等的时间太久、业务员态度不好等。服务态度不佳的情况还有:服务热线过“热”,很难打进去,接电话的态度有时候恶劣,对于事故采用回避的处理方式。
2.3.5 重金属 取“2.3.4”项下遗留的残渣适量,依2010年版《中国药典》(二部)附录ⅧH第二法检查。结果,6批样品重金属的质量分数均不超过百万分之十。
2.3.6 水分和干燥失重 取本品适量,分别依2010年版《中国药典》(二部)附录ⅧM第一法水分测定法和附录ⅧL干燥失重法测定,结果显示两种方法结果无显著性差异。由于采用60 ℃减压干燥的方法,操作更简单快捷,所以拟定采用60 ℃减压干燥至恒质量的方法来测定样品的水分。由于本品含半分子结晶水[3],按理论值计算含水量应为4.6%,考虑样品的半分子结晶水和储存过程中的吸附水,将其限度定为:干燥减失重应为4.0%~6.0%。
2.3.7 有关物质 采用HPLC法对有关物质进行考察。现行的质量标准中均收载有此检查项,但色谱条件不尽相同,并且由于本品是以2-甲基-5-硝基咪唑作为起始原料,现行的标准均未对2-甲基-5-硝基咪唑进行控制,不利于控制产品质量[4-5],因此有必要对其标准进行研究。
(1)方法的确定。依照2010年版《中国药典》(二部)附录ⅤD HPLC法测定[6],用十八烷基硅烷键合硅胶为填充剂,以甲醇-水(20∶80,V/V)为流动相,检测波长为318 nm。理论板数按塞克硝唑峰计算不低于2 000,塞克硝唑峰与相邻杂质峰的分离度应符合要求。取本品适量,加流动相溶解并稀释制成1 ml中约含0.3 mg的溶液,作为供试品溶液;另取2-甲基-5-硝基咪唑对照品约15 mg,精密称定,置于100 ml量瓶中,加流动相溶解并稀释至刻度,摇匀,精密量取2 ml,置于10 ml量瓶中,加流动相稀释至刻度,摇匀,作为对照品贮备液。分别精密量取供试品溶液1 ml与对照品贮备液1 ml,置于同一100 ml量瓶中,用流动相稀释至刻度,摇匀,作为对照品溶液。精密量取对照品溶液20 μl注入HPLC仪,调节检测灵敏度,使塞克硝唑色谱峰的峰高为满量程的10%~20%,再精密量取供试品溶液及对照品溶液各20 μl,分别注入HPLC仪,记录色谱图至主成分峰保留时间的2倍。
(2)样品有关物质检查。取本品适量,按上述色谱条件检查有关物质,结果见表1。
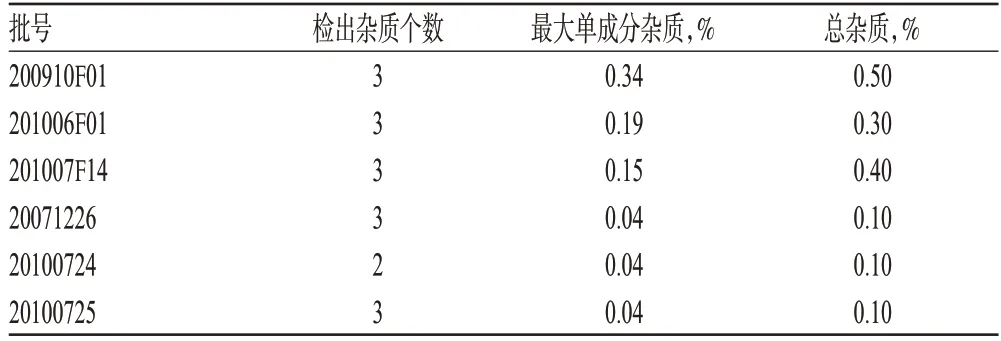
表1 有关物质检查结果Tab 1 Results of related substances
2.3.8 残留溶剂 由于原料生产工艺中可能使用了乙酸乙酯、二氯甲烷、甲苯,所以本研究对这3种残留溶剂进行了考察。
(1)方法的确定。取本品适量,精密称定,加二甲亚砜稀释制成每1 ml中约含20 mg的溶液,作为供试品溶液;另取甲苯、乙酸乙酯、二氯甲烷各适量,精密称定,加二甲亚砜溶解,制成每1 ml中约含甲苯0.017 8 mg、乙酸乙酯0.1 mg和二氯甲烷0.012 mg的溶液,作为对照品溶液。依2010年版《中国药典》(二部)附录ⅧP第三法残留溶剂法测定,以6%氰丙基苯基-94%二甲基聚硅氧烷作为固定液;起始柱温90 ℃,维持5 min,以每分钟20 ℃的速率升温至170 ℃,维持5 min;检测器温度为200 ℃;进样口温度为220 ℃。甲苯、乙酸乙酯、二氯甲烷的分离度应符合要求。精密量取供试品和对照品溶液各0.5 μl,注入GC仪,按外标法以峰面积计算,应符合要求。
(2)样品残留溶剂检查。取本品适量,按上述方法检查残留溶剂,结果均未检出甲苯、乙酸乙酯、二氯甲烷。
2.4 含量测定
由于本品分子结构中含有咪唑环,依据原料药首选容量分析法测定含量的原则[7-8],采用非水滴定法测定含量。由于现行质量标准中所选溶剂和指示剂不尽相同,故有必要对其进行研究和统一。
2.4.1 溶剂和指示剂的选择 取本品(厂家:A公司,批号:201006F01)约0.13 g,共8份,其中2份加入冰醋酸20 ml,2份加入冰醋酸10 ml+醋酐10 ml、2份加入冰醋酸5 ml+醋酐15 ml、2份加入醋酐20 ml使溶解。在同一溶剂溶解的2份样品中,分别加入结晶紫指示液及萘酚苯甲醇指示液,均用高氯酸滴定液(0.1 mol/L)滴定,用电位滴定法指示终点。结果表明,采用冰醋酸20 ml作为溶剂时,滴定终点突跃明显;采用结晶紫作为指示液时,突跃点时溶液的颜色突变为亮绿色;采用萘酚苯甲醇作为指示液时,突跃点时溶液的颜色突变为亮绿色,两者无显著性差异。考虑到检验的经济性,选择结晶紫作为指示剂。
2.4.2 方法的确定 取本品约0.13 g,加入冰醋酸20 ml使溶解,加入结晶紫指示液1滴,用高氯酸滴定液(0.1 mol/L)滴定至溶液现亮绿色,并将滴定的结果用空白试验校正。每1 ml的高氯酸滴定液(0.1 mol/L)相当于18.52 mg的C7H11N3O3。电位滴定曲线详见图1(样品批号:201006F01)。
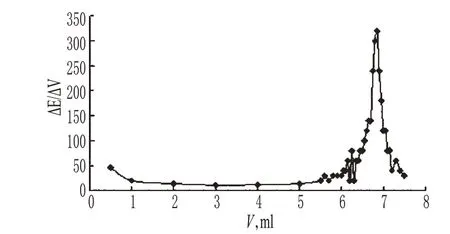
图1 电位滴定曲线Fig 1 The profile of the potentiometric titration
2.4.3 电位滴定法与容量分析法的比较 取本品(A公司,批号:201006F01)约0.13 g,加入冰醋酸20 ml使溶解,用高氯酸滴定液(0.1 mol/L)滴定,分别采用电位滴定法和容量分析法(结晶紫为指示液)进行测定。结果,两种方法测定结果无显著性差异,详见表2。

表2 含量测定方法结果比较(%)Tab 2 Results of content determination(%)
2.4.4 回收率试验 精密称取本品(A公司,批号:201006F01)约0.052、0.065、0.078 g,各3份,分别置于锥形瓶中;另精密加入60 ℃减压干燥至恒质量的塞克硝唑对照品约0.052、0.065、0.078 g,各3份,分别加入冰醋酸20 ml溶解后,加结晶紫指示液1滴,用高氯酸滴定液(0.1 mol/L)滴定至溶液现亮绿色,并将滴定结果用空白试验校正。每1 ml的高氯酸滴定液(0.1 mol/L)相当于18.52 mg的C7H11N3O3,结果见表3。
2.4.5 重复性试验 取本品约0.13 g(A公司,批号:201006F01),共6份,精密称定,加入冰醋酸20 ml溶解后,加结晶紫指示液1滴,用高氯酸滴定液(0.1 mol/L)滴定至溶液现亮绿色,并将滴定结果用空白试验校正。每1 ml的高氯酸滴定液(0.1 mol/L)相当于18.52 mg的C7H11N3O3。结果,RSD=0.42%(n=6),表明该方法重复性良好。
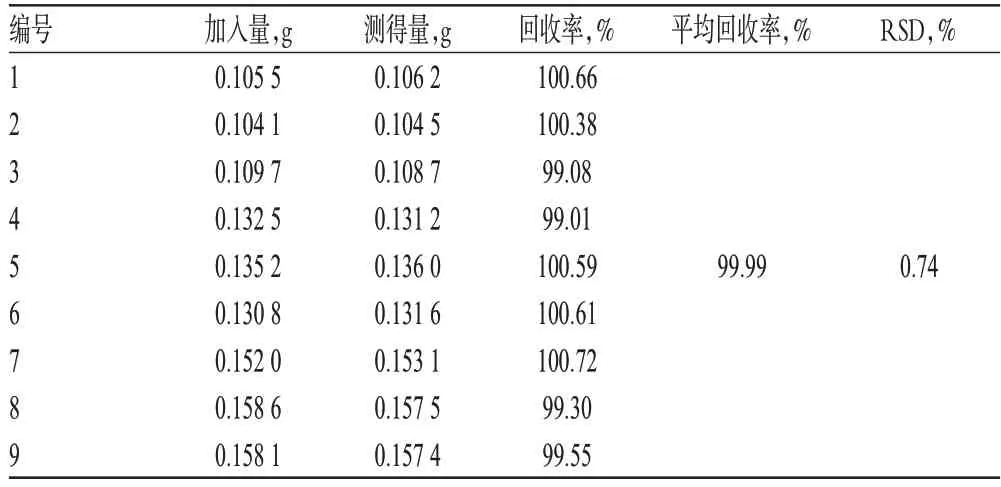
表3 回收率试验结果Tab 3 Results of recovery rates
2.4.6 样品含量测定 样品含量测定结果详见表4。

表4 样品含量测定结果(%)Tab 4 Results of sample’s content(%)
3 讨论
塞克硝唑是硝基咪唑类化合物,与甲硝唑等传统的硝唑类药物有相似的化学结构,具有生理活性更强、不良反应少、半衰期长等特点[9]。本文统一了其性状、鉴别、检查与含量测定方法,可为塞克硝唑质量标准的修订提供依据。
[1]Rossignol JF,Maisonneuve H,Cho YW.Nitroimidazoles in the treatment of trichomoniasis,giardiasis and amebiasis[J].Int J Clin Pharmacol Ther Toxicol,1984,22(2):63.
[2]Edwards DI.Nitroimidazole drugs-action and resistance mechanisms:mechanisms of action[J].J Antimicrob Chemother,1993,31(1):9.
[3]向红琳,胡高云,徐康平,等.塞克硝唑1/2水合物的研究[J].中南药学,2004,2(2):85.
[4]张锦琳,袁耀佐,钱文,等.HPLC法测定塞克硝唑的含量及有关物质[J].中国药科大学学报,2009,40(6):527.
[5]孟婷,赵敏,吕韶敏,等.高效液相色谱法测定塞克硝唑中有关物质[J].第四军医大学学报,2007,28(3):282.
[6]国家药典委员会.中华人民共和国药典:二部[S].2010年版.北京:中国医药科技出版社,2010:附录ⅤD.
[7]杭太俊.药物分析[M].北京:人民卫生出版社,2012:143.
[8]申献玲.塞克硝唑温敏性水凝胶的处方设计与含量测定[J].中国药房,2010,21(9):840.
[9]Nunez JT,Gomez M.Lowdosesecnidazole in the treatment of bacterial vaginosis[J].Int J Gynaecolobstet,2005,88(3):281.
猜你喜欢
商业评论(2022年4期)2022-05-05
化学教学(2020年4期)2020-05-13
中国现代医生(2019年24期)2019-11-23
中国现代医生(2018年23期)2018-12-18
科学与财富(2018年26期)2018-10-24
中国民族民间医药·上半月(2018年1期)2018-09-18
中国科技纵横(2018年10期)2018-07-27
上海故事(2015年5期)2015-04-17
舰船电子工程(2012年8期)2012-07-11
意林(2011年6期)2011-05-14
