
椒江口沉积物中细菌多样性初步研究*
2015-03-08 05:27王健鑫郑俊威俞凯成刘明华
海洋与湖沼 2015年4期
蒋 然 王健鑫① 黄 备 张 潘 郑俊威 俞凯成 刘明华
(1. 浙江海洋学院 海洋微生物分子生态与应用实验室 舟山 316022; 2. 浙江省舟山海洋生态环境监测站 舟山 316021)
微生物是生态系统中的生产者、消费者,也是分解者,在维持环境稳定的过程中起着动植物无法取代的作用(杜萍等,2012)。同时,由于微生物对环境变化十分敏感,其多样性及群落结构常作为环境变化的指示因子(王金成等,2012; Zhuet al,2013)。
椒江是浙江省第三大河流,入台州湾,其盐淡水以强混合型为主,为典型的山溪性强潮河口,沉积物主要来自海域沙泥(谢钦春等,1998)。由于椒江口上游是化工、制药、火力发电等企业的所在地,水体中已发现多溴联苯醚(PBDEs)、滴滴涕(DDTs)、六六六(HCHs)、重金属等多种污染物(江锦花等,2006; 余鹏等,2011; 杨华云等,2014),沉积物作为承载各种污染物的载体,其相关生物更是受到巨大的环境压力。
关于椒江口生物与生态的研究起初关注较多的是大型底栖动物资源及鱼类养殖环境调查,后来陆续开展了对虾蟹、浮游动物等方面的研究(江锦花等,2007;赵永强等,2009; 杜萍等,2011a; 齐海明等,2013)。但对于包含巨大环境信息的微生物群落结构的研究,还比较欠缺: 杜萍等(2011b)首先对椒江口春季水体中异氧细菌及氮、磷细菌的生态分布特征进行了研究,其后又采用Biolog和PCR-DGGE技术对椒江口沉积物微生物多样性进行了分析(杜萍等,2012),但主要侧重于站点的多样性指数及站点与石油烃的关系,并没有对具体的细菌类群进行讨论; Hu等(2012)分析了椒江口厌氧氨氧化细菌的多样性与化学污染物(硝基苯,甲苯,多环芳烃等)的关系; Shen等(2014)采用分子生态学方法对椒江口氮依赖型厌氧甲烷氧化细菌进行了研究,上述两个研究则是针对特殊的细菌种群,没有涉及群落中其它类别细菌的多样性及结构变化的相关信息。
本文采用常规生理生化及分子鉴定的方法对椒江口沉积物中可培养细菌的多样性进行分析,并采用构建环境总 DNA克隆文库方法,对未培养细菌的多样性及系统发育进行研究,以期为椒江口沉积环境生态的相关研究提供科学参考。
1 材料与方法
1.1 样品采集
2013年5月23日,在椒江口化工园区附近选择C0 (28°41'27.6"N,121°26'49.2"E)、C1 (28°41'6"N,121°29'13.2"E)、C2 (28°40'32.16"N,121°33'32.4"E)3个站点(图 1),每个站点采集表层沉积物样品一份,在无菌条件下分装于严格灭菌的样品瓶内,-20°C保存,后置于实验室-80°C超低温冰箱中长期保存。
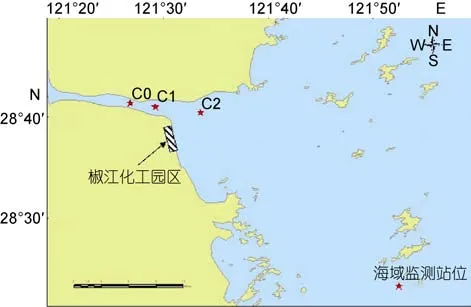
图1 椒江口采样站点Fig.1 Sampling station in the Jiaojiang River estuary
1.2 沉积物环境化学指标测量
常规水化学指标的测定参照海底沉积物化学分析方法(Heijset al,2008); 总有机碳含量(TOC)用总有机碳分析仪(TOC-V CSH,日本岛津)进行测定; 总氮含量(TN)用蛋白质自动分析仪(SKALAR Primacs SN,荷兰)进行测定; 总磷含量(TP)用钼酸铵分光光度法进行测定(国标GB/T20260-200)。
1.3 可培养细菌的分离和鉴定
1.3.1 细菌分离纯化 称取10.0 g沉积物样品,放入盛有90 mL已灭菌海水的锥形瓶(含玻璃珠)中,180 r/min震荡 30 min。静置使其自然沉降,取上清稀释1000倍,涂布于Zobell 2216培养基,置于25°C培养,待长出明显菌落后挑单菌落并反复划线纯化菌株。
1.3.2 细菌形态学鉴定 对分离纯化得到的菌株,首先观察并记录菌落形态,进行革兰氏染色,在确定是否生成芽孢之后,进行下列实验(李振高等,2008):(1) 革兰氏阳性芽孢杆菌: 测定菌株与氧的关系; (2)革兰氏阳性无芽孢杆菌: 镜检菌体形态及其随培养时间的变化; (3) 革兰氏阴性无芽孢杆菌: 进行氧化酶实验; (4) 菌体形态是球形者,要观察其幼龄及老龄菌体形态,确定其没有杆状菌。
1.3.3 细菌生理生化鉴定 由于海洋细菌中革兰氏阴性菌远多于革兰氏阳性菌,因此对所得菌株采用法国生物梅里埃公司的 API-20E(肠杆菌和其它革兰阴性杆菌科鉴定系统)、API-20NE(非肠道革兰阴性杆菌科鉴定系统)进行鉴定。API-20E和API-20NE能对需鉴定菌株进行23个生化试验,并以其自身代谢产物的颜色变化或加入试剂后的颜色变化加以鉴定,结果进入数据库检索后得到相应的种名(黄备等,2009)。
1.3.4 细菌16S rDNA基因扩增及鉴定 选取典型可培养菌株,用细菌基因组 DNA提取试剂盒(TaKaRa公司)获得菌株基因组 DNA,采用细菌 16S rDNA通用引物 27F(5'-AGAGTTTGATCCTGGCTCA G-3')和1492R(5'-TACGGTTACCTTGTTACGACTT-3')进行PCR扩增。50 μL反应体系为: 10×buffer 5 μL,dNTPs 0.5 μL,Primer 27F 1μL,Primer 1492R 1 μL,H2O 41 μL,Taq 酶(TaKaRa) 0.5 μL,模板 DNA 1 μL。PCR扩增条件为: 95°C预变性5 min; 94°C变性1 min,56°C退火30 s,72°C延伸2 min,循环35次; 最后72°C延伸10 min。PCR产物送上海美吉生物技术有限公司纯化并进行DNA序列测定。选取长度超过1300 bp的序列进行 BLASTn(www.ncbi.nlm.nih.gov/BLAST/)比对,获得相似性大于97%的同源类群序列信息。
1.4 未培养细菌多样性研究
1.4.1 环境总DNA提取和16S rDNA扩增 利用FastPrep快速核酸提取仪(MP Biomedicals公司)和FastDNA spin kit for soil试剂盒进行沉积物DNA提取,核酸蛋白检测仪(Bio-Rad公司)测定DNA浓度和纯度,纯化后用于PCR扩增。扩增采用引物341F(5'-CCTA CGGGAGGCAGCAG-3')和907R(5'-CCGTCAATTCC TTTGAGTTT-3')。扩增反应体系(条件)与可培养细菌分子鉴定相同。
1.4.2 未培养细菌克隆文库构建及序列分析 PCR回收产物与pMD-18T vector (TaKaRa公司)在16°C连接过夜,将连接产物转化到大肠杆菌(E. coli)DH5α感受态细胞(TaKaRa公司),经蓝白斑筛选,挑选阳性克隆,重新扩增插入片断,将含有合适大小插入片段的克隆送上海美吉生物技术有限公司进行测序。
获得序列后,首先利用Bellerophon (http://compbio.anu.edu.au/bellerophon/bellerophon.pl)在线去除嵌合体; 再应用BLASTn程序搜索相似性序列,进行系统发育分析。采用ClustalX (Version 1.8)对序列进行比对分析,通过 MEGA 5软件构建系统发育树(Kumaret al,2004),采用Neighbor-Joining建树方法,建树结果进行 Bootstrap1000次系统检验; 利用PHYLIP软件包中DNASIS程序计算距离矩阵,利用DOTUR软件将相似性>97%的序列归为一个 OTU,计算多样性指数并绘制稀释曲线(Schlosset al,2005)。未培养菌株16S rDNA的序列登录号为: C0:KP016015—KP016127; C1: KP016128—KP016210;C2: KP016211—KP016297; 可培养菌株 16S rDNA的序列登录号为: KP016596—KP016699。
2 结果与分析
2.1 样品环境化学指标
椒江口C0、C1、C2站点沉积物样品的环境化学相关指标如表1所示,从表1中可知,三个站点中C0的总有机碳(TOC)含量(6.74 g/kg)和总氮(TN)含量(528 mg/kg)最高,各站点间TOC和TN含量由高到低依次为为: C0>C2>C1; 而总磷(TP)含量最高站点为 C2(628mg/kg),TP含量在三个站点排序为: C2>C1>C0。
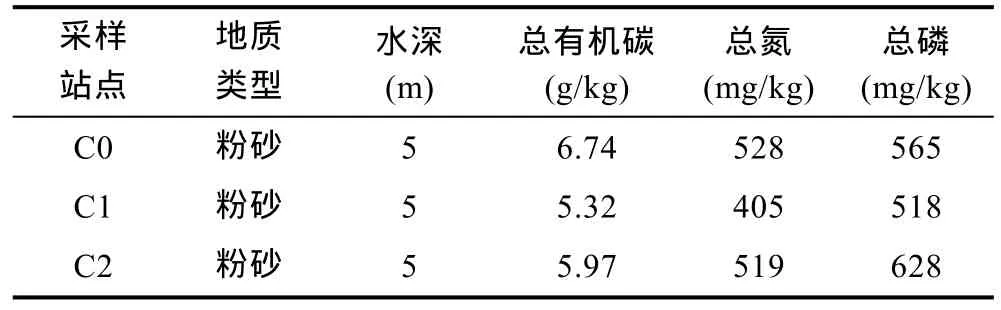
表1 椒江口沉积物环境化学指标Tab.1 Chemical measurements of sediment in Jiaojiang River estuary
2.2 纯培养菌株的生理生化鉴定结果
分离纯化得到 78株可培养细菌,其中革兰氏阴性菌占 66.7%,革兰氏阳性菌占 33.3%。细菌菌落绝大多数呈乳白色不透明、边缘整齐光滑的形态,显微镜下细菌形状以杆状居多,大多数氧化酶反应呈阳性。根据菌落形态和菌体基本特征,我们挑选其中19株典型的革兰氏阴性菌进行API-20NE及API-20E鉴定,结果如表2所示。细菌皆属于变形菌门,其中52.6%为杀鲑气单胞菌杀鲑亚种(Aeromonas salmonicidasp.salmonicida),远大于象山港等地海水样品中所鉴定出的16.8%(黄备等,2009); 其余菌株所占比例较小,创伤弧菌(Vibrio vulnificus)、溶藻胶弧菌(Vibrio alginolyticus)以及洋葱假单胞菌(Pseudomonas cepacia)各占 10.5%,产碱普罗威登斯菌(Providencia alcalifaciens)、塔特姆氏菌(Tatumella ptyseos)、少动鞘氨醇单胞菌(Sphingomonas paucimobilis)各占5.3%。
2.3 纯培养菌株16S rDNA分子鉴定
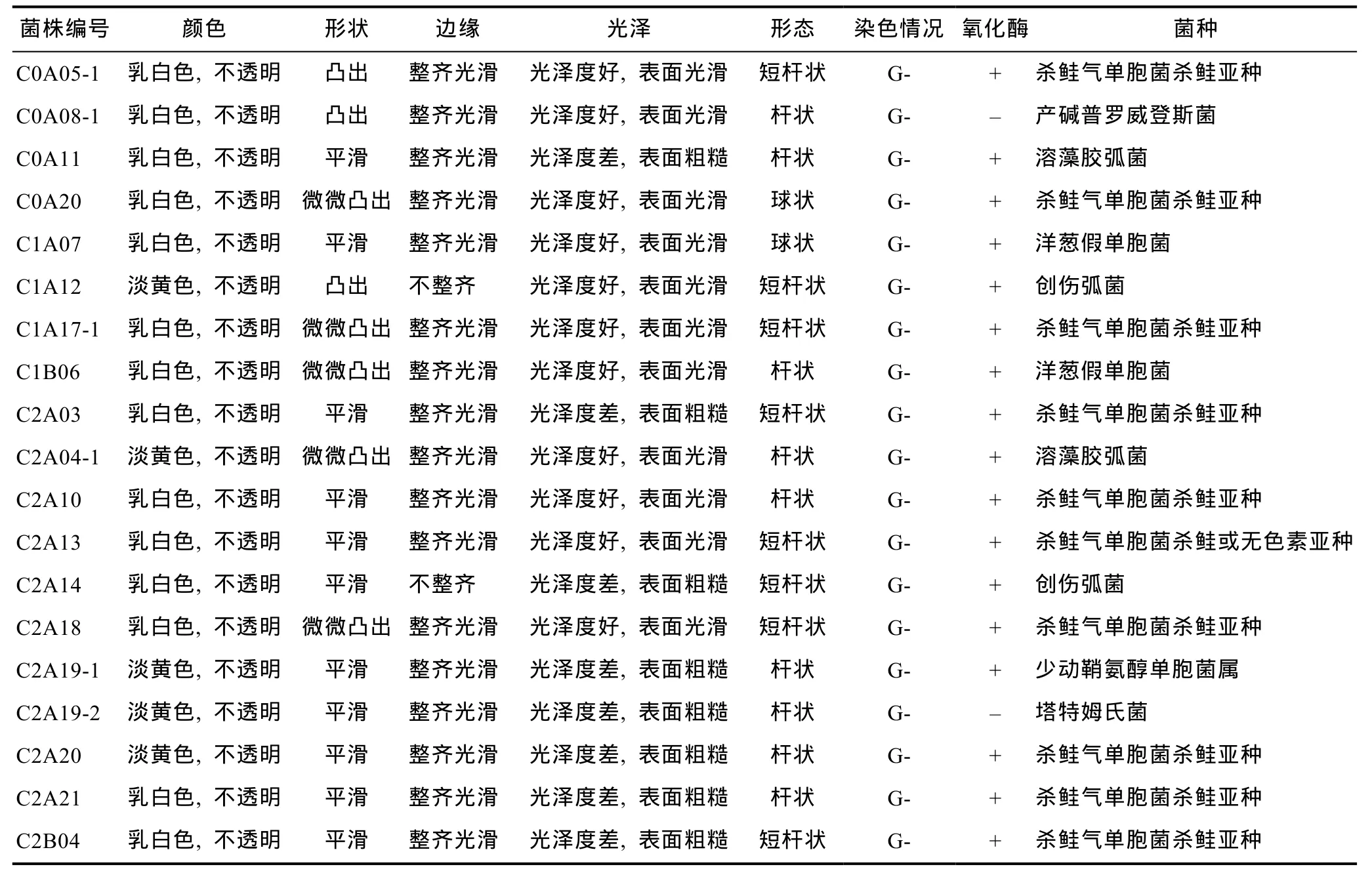
表2 典型可培养细菌常规鉴定结果Tab.2 Identifications of the culturable bacteria
选取43株菌株进行16S rDNA序列测定,得到41个片段长度为 1400bp左右的有效序列。通过BLASTn将所得序列进行比对,41个克隆子主要属于4 个门类: 厚壁菌门(Firmicutes)、γ-变形菌纲(Gammaproteobacteria)、放线菌门(Actinobacteria)以及 β-变形菌纲(Betaproteobacteria),各站点克隆文库中不同类群细菌的比例如图2所示。
三个站点中均有弧菌(Vibrio)、海洋芽孢杆菌(Oceanobacillus)、芽孢杆菌(Bacillus)的分布,其中弧菌在每个站点的比例都超过50%,为优势菌种。相对于C0和C2而言,C1的多样性更丰富,分布有其它站点没有的类群,如: 产碱杆菌(Alcaligenes)、赖氨酸芽孢杆菌(Lysinibacillus)、节杆菌(Arthrobacter),但在C0和 C2站点也发现了 C1站点没有的微小杆菌(Exiguobacterium),至于芽胞八叠球菌(Sporosarcina)则只在C2站点存在。
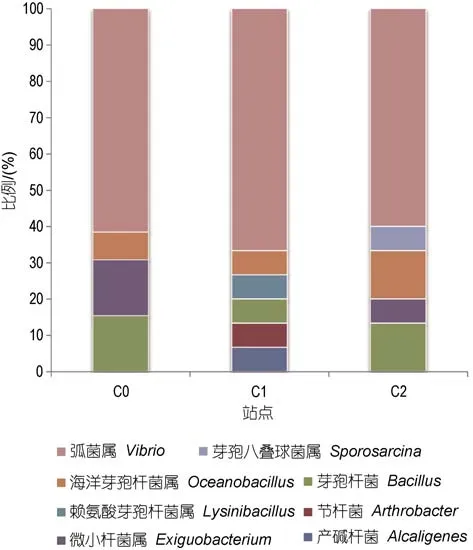
图2 三个站点克隆文库的类群比例Fig.2 The ratios of the clone libraries in taxonomic group of the three stations
利用 DOTUR将相似性>97%的序列归为一个OTU,41个克隆子共有14个OTU。并对所得OTUs进行16S rDNA系统发育树的构建(如图3所示)。由系统发育树结果可知,厚壁菌门、芽孢杆菌纲、芽孢杆菌目的丰度和多样性最为丰富(占 31.7%),其中芽孢杆菌属有巨大芽胞杆菌(Bacillus megaterium)、黄海芽胞杆菌(Bacillus marisflavi)、短小芽胞杆菌(Bacillus pumilus)等种类; 另外还含有海洋芽孢杆菌属(Oceanobacillus)、芽胞八叠球菌属(Sporosarcina)、微小杆菌属(Exiguobacterium)。γ-变形菌纲多样性仅次于厚壁菌门,其基因序列都为弧菌属,包括沙蚕弧菌(Vibrio nereis)、溶藻胶弧菌(Vibrio alginolyticus)、副溶血性弧菌(Vibrio parahaemolyticus),其中C1B06的基因序列(含 23个菌株且在 3个站点均匀分布)与2007年江苏暴发的大规模蛤蜊死亡有关的副溶血弧菌MM21有高同源性(Yueet al,2010)。β-变形菌门以及放线菌门仅有一个 OTU,分别属于粪产碱菌(Alcaligenes faecalis)、原玻璃蝇节杆菌(Arthrobacter protophormiae)。
2.4 非培养细菌多样性分析
2.4.1 非培养细菌16S rDNA文库分析 每个站点选取 120个左右含有正确插入片段的细菌进行测序,返回的序列首先经过 sequin软件去除载体,再通过Bellerophon网站去掉嵌合体,3个站点最终分别获得113、84、89个有效克隆子,片段长度为550—700bp。
利用 DOTUR软件,将相似性>97%的克隆子归为一个OTU,C0、C1和C2三个站点的OUT分别为59、57和72个。三个站点克隆文库的多样性指数如表3所示,C0、C1、C2三个站点Shannon指数依次递增,Simpson指数依次递减,说明C2站点的文库多样性最高,C1站点次之,C0站点最低,这与稀释性曲线(图4)所呈现的结果一致。从Chao1指数和Jack指数来看,C1站点拥有最高的物种丰富度(Richness)。导致丰富度指数与多样性指数结果不一致的原因可能是多样性指数除了受丰富度影响,还受到均匀度(Aveargeness)影响(Magurran,2011)。

表3 椒江口沉积物细菌多样性指数一览表Tab.3 Bacterial diversity indices in sediment of the Jiaojiang River estuary
2.4.2 三个站点克隆文库物种类群分析 通过BLASTn搜索相似序列可将3个站点共286个克隆子可分为8个门(群),分别为变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)、绿弯菌门(Chloroflexi)、芽单胞菌门(Gemmatimonadetes)、硝化螺旋菌门(Nitrospirae)、CFB 群(Cytophaga-Flavobacterium-Bacterioides)、放线菌门(Actinobacteria)、厚壁菌门(Firmicutes); 另有 23.1%的细菌分类地位不确定; 这些相似序列主要来自于海洋或河流沉积物、海水或河水,少数来自土壤。

图3 根据可培养细菌16S rDNA基因序列构建的系统发育树Fig.3 Phylogenetic tree of the culturable bacteria in 16S rDNA gene sequence

图4 三个文库的稀释性曲线Fig.4 The rarefaction curve of three clone libraries
由图5可看出: C0站点文库拥有最多类群,主要由变形菌门及绿弯菌门构成,分别占 37.17%和26.55%,绿弯菌含量为3个站点中最高; 变形菌门以γ-变形菌纲为主要类群,占文库的21.24%,明显高于其余两个站点γ-变形菌含量; 其次为α-变形菌纲,占10.62%,另有少量的 δ-变形菌纲和 β-变形菌纲。C1站点文库所含类群最少,变形菌门为其最主要类群(占 60.71%)。变形菌门中 α-、β-、δ-所占比例为三个站点中最高,分别为23.81%、11.90%和11.90%。C2站点文库同样以变形菌门为主要类群,但其所占比例仅为32.59%,放线菌门也是C2站点文库的优势类群,占 17.98%,远高于其余两个站点的放线菌含量;同时值得注意的是,该站点分类地位未确定的细菌所占比例在3个站点中最高(33.71%)。
综合3个站点文库,变形菌门为最大优势菌群占42.7%,其中 γ-变形菌占 16.8%,α-变形菌占 14.7%,δ-变形菌占6.3%,β-变形菌占4.9%,这与珠江及长江口沉积物中优势菌群为 δ-变形菌有明显差异(姜丽晶,2007; 王新新,2008); 仅次于变形菌门的是绿弯菌门(占 11.5%),这与西沙海槽表层沉积物中绿弯菌门所占比例(8.5%)相似(Liet al,2008); 第三大优势菌群为放线菌门(占10.5%)。酸杆菌、变形菌、放线菌、CFB群在3个站点均有出现,但含量不高; 芽单胞菌只出现在C0站点; CFB group中的Latescibacteria只在C2站点中出现。
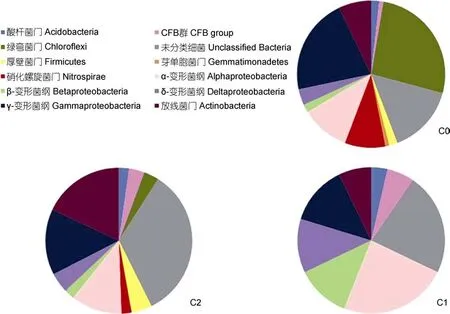
图5 三个站点克隆文库的类群比例Fig.5 The clone librariy ratios of the bacterial groups from the three stations
2.5 非培养细菌系统发育分析
2.5.1 C0站点非培养细菌系统发育分析 使用MEGA5.0软件,将C0站点典型菌BLASTn的比对结果构建系统发育树,结果如图6。
由系统发育树结果可知,α-变形菌纲中在 C0站点多样性最为丰富,基因型C0_328(2个克隆子)与活性污泥中的未培养生丝微菌科(Hyphomicrobiaceae)克隆子DMSN111有高同源性(98%),与基因型C0_306(2个克隆子)有高同源性的相似序列则是分离自海水、与降解溴化甲烷有关的的可培养鞘氨醇单胞菌(Sphingomonadaceae Oxy6),基因型 C0_10(1个克隆子)的相似序列为美国墨西哥湾深水地平线事故区域的未培养α-变形菌克隆子S2-8-036,基因型C0_29(1个克隆子)与被脂肪族烃污染的土壤中的生丝微菌的克隆子(Hyphomicrobiumsp.CM38D10)有较高同源性(98%)。γ-变形菌纲在C0站点也有较多分布,基因型C0_69(含14个克隆子)与台湾淡水河河口的未培养鱼立克次体科的克隆子(Piscirickettsiaceae DS021)有较高同源性(97%),基因型 C0_323(2个克隆子)与被石油污染的西班牙 Cies群岛潮下带沉积物中未培养 γ-变形菌克隆子 RODAS-143有高同源性(99%),基因型 C0_44(1个克隆子)的序列与重金属污染的比利时大陆板块沉积物中未培养 γ-变形菌克隆子 beligica 2005/10-ZG-6相似度最高。δ-变形菌纲中基因型C0_6(2个克隆子)的相似序列为脂肪烃污染土壤中的未培养δ-变形菌克隆子CMOC10。
虽然绿弯菌门克隆子在C0站点总克隆子中所占比例高于变形菌门中的α-、γ-变形菌纲,但其多样性远不如以上两种类群,这主要是由于绿弯菌门中克隆子大多归属于 C0_129基因型(24个克隆子),它与法国南部工业区被原油污染的Etang de Berre潟湖沉积物中的未培养绿弯菌克隆子17bis T0h-oil同源性为96%。C0_42(2个克隆子)的相似序列为法国 Guiana海岸与二氧化碳固定有关的未培养绿弯菌克隆子4_211。
硝化螺旋菌是一类可以将亚硝酸盐氧化成硝酸盐的细菌(李海艳,2009),在海洋氮循环里占据重要位置。C0站点的基因型 C0_128(9个克隆子)属于硝化螺旋菌,与表层东海沉积物中的未培养硝化螺旋菌DH132B19有高同源性(98%),基因型C0_47(1个克隆子)的相似序列为德国Düsseldorf市Flingern地区被石油污染的沉积物中未培养硝化螺旋菌克隆子D15_16。
C0站点还含有一定数量的放线菌,共 8个克隆子分属于4个OTU,其中基因型C0_88(5个克隆子)的相似序列为印度坎贝湾(临近化工城市巴罗达)海滨土壤中的未培养放线菌 ONGS217,剩余 3个克隆子分属于3个不同的OUT,其相似序列都来自污染环境,基因型 C0_1相似序列为德国 Düsseldorf市 Flingern地区被石油污染的沉积物中的红色杆菌(Rubrobacteraceaesp. D15_40),基因型C0_332(1个克隆子)和基因型C0_334(1个克隆子)的相似序列为石油污染土壤中的两个放线菌克隆子EK_CK647和EK_CK623。
在分类地位不确定的细菌中克隆子中,基因型C0_336(1个克隆子)与PAH污染的土壤中的未培养细菌克隆子PYR10d12有高同源性(99%)。
2.5.2 C1站点非培养细菌系统发育分析 使用MEGA5.0软件,将C1站点典型菌BLASTn的比对结果构建系统发育树,结果如图7。
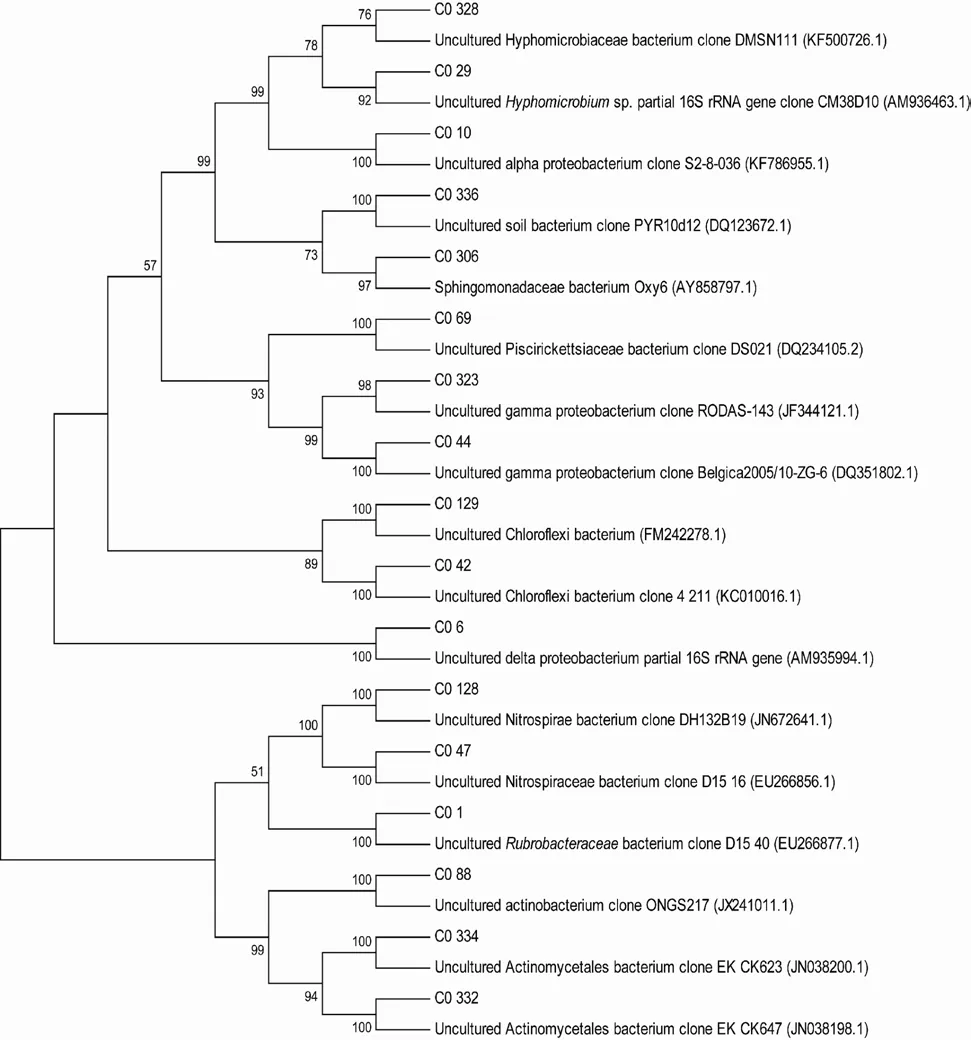
图6 C0站点根据细菌16S rDNA基因序列构建的系统发育树Fig.6 Phylogenetic tree of bacteria in 16S rDNA gene sequences in Station C0
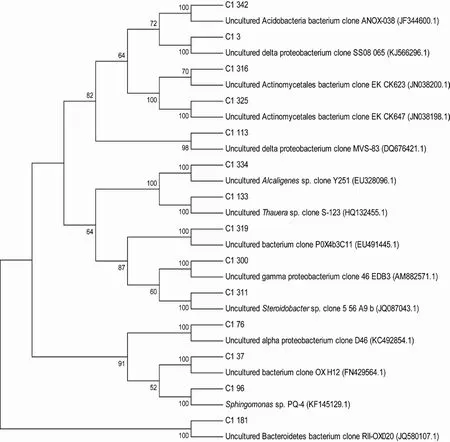
图7 C1站点根据细菌16S rDNA基因序列构建的系统发育树Fig.7 Phylogenetic tree of bacteria in 16S rDNA gene sequence in Station C1
由系统发育树的结果可知,C1站点克隆文库最主要类群依然为变形菌门中的 α-变形菌纲和 γ-变形菌纲。α-变形菌纲中,基因型C1_76(6个克隆子)相似序列为波罗的海及黑海中未培养 α-变形菌的克隆子D46,基因型C1_96(3个克隆子)相似序列为浙江水环境(湿地、红树林、海洋)沉积泥中能降解菲或芘以及能产 AHL的可培养鞘氨醇单胞菌(Sphingomonassp.PQ-4)。γ-变形菌纲中,基因型C1_300(1个克隆子)和C1_311(1个克隆子)的相似序列都来自被石油烃污染的区域,前者为Etang de Berre潟湖海岸沉积物的γ-变形菌克隆子46 EDB3,后者为含水层的γ-变形菌克隆子 5_56_A9_b。C1文库中 β-变形菌和 δ-变形菌所占比例明显高于其它两个站点。δ-变形菌纲中,基因型C1_113(4个克隆子)与来自高有机物输入的低氧缺氧淡水沉积物中的未培养δ-变形菌克隆子MVS-83同源性为94%,基因型C1_3(3个克隆子)相似序列为北极Svalbard海湾的未培养δ-变形菌克隆子SS08_065。β-变形菌纲中基因型C1_133(6个克隆子)与湘江重金属污染区域沉积物中所检测到的未培养陶厄氏菌(Thauerasp. clone S-123)有高同源性(98%),基因型C1_334(1个克隆子)相似序列为原油污染的土壤中的产碱杆菌克隆子(Alcaligenessp. Clone Y251)。
C1站点还有一些其它基因型,C1_181基因型(4个克隆子)与石油污染的西班牙Cies群岛的潮下带沉积物的未培养拟杆菌克隆子 RII-OX020有高同源性(98%); 基因型 C1_342(1个克隆子)的相似序列为未培养酸杆菌门克隆子ANOX-038(来源环境与C1_181一样); 基因型C1_316(1个克隆子)和C1_325(1个克隆子)的相似序列为石油污染土壤的未培养放线菌克隆子EK_CK623和EK_CK647。
在 C1站点分类地位未明确的细菌中,基因型C1_37(1个克隆子)的相似序列为法国Etang de Berre石油化工厂沉淀池中的未培养细菌克隆子 OX H12;基因型C1_319(1个克隆子)相似序列为美国北卡罗来纳州工业煤气厂的 PAH污染土壤中未培养细菌的克隆子P0X4b3C11。
2.5.3 C2站点非培养细菌系统发育分析 使用MEGA5.0软件,将C2站点典型菌BLASTn的比对结果构建系统发育树,结果如图8。
由系统发育树结果可知,C2站点主要优势种群同样为变形菌门的α-变形菌及γ-变形菌。γ-变形菌纲中,基因型C2_31(3个克隆子)与突尼斯Bizerte地区潟湖沉积物中的未培养 γ-变形菌克隆子 5m-84同源性为 95%,C2_337(2个克隆子)与重金属污染的比利时大陆板块沉积物中的未培养 γ-变形菌克隆子Belgica2005/10-130-24有高同源性(100%),C2_310(2个克隆子)与南大洋中与铁肥效应有关的未培养鱼立克次体科(Piscirickettsiaceae)克隆子C146300241有较高同源性(97%),基因型C2_29(1个克隆子)的相似序列为印度临近化工城市巴罗达的坎贝湾的海滨土壤的未培γ-变形菌克隆子 ONGS147。α-变形菌纲中,基因型C2_2(1个克隆子)相似序列为南京铅锌矿残渣污染的土壤中生丝微菌科(Hyphomicrobiaceae)克隆子H3-43,基因型 C2_313(1个克隆子)与污水处理厂气溶胶中的Catellibacteriumsp. clone OTU-27-40m有高同源性(99%),基因型 C2_320(1个克隆子)与可降解原油的Altererythrobactersp. clone OB38-2有高同源性。β-变形菌纲中,基因型 C2_300的相似序列为湘江重金属污染区域沉积物检测到的未培养陶厄氏菌(Thauerasp. clone S-123)。δ-变形菌纲中,基因型C2_358(1个克隆子)与被原油污染的地中海 Mallorca岛海岸沉积泥中未培养δ-变形菌克隆子MS-A223有较高同源性(98%)。
放线菌门为污染环境中十分活跃的类群(Nogaleset al,2001),在C2站点文库中分布比较丰富。基因型C2_317(2个克隆子)相似序列为东海大陆架表层沉积物中的未培养放线菌克隆子 DH133B17; 基因型C2_27(2个克隆子)与浙江东阳填埋场沉积物中的放线菌克隆子 De371有较高同源性(97%); 基因型C2_333、C2_336和C2_352(均为1个克隆子)的相似序列来自同一个石油污染土壤环境,分别为未培养放线菌目克隆子 Actinomycetales bacterium clone EK_CK623、EK_CK647和 EK_CK647; 基因型C2_374(1个克隆子)和C2_72(1个克隆子)相似序列都来自印度临近化工城市巴罗达的坎贝湾的海滨土壤,分别为未培养放线菌克隆子ONGS161、ONGS217。
酸杆菌门和拟杆菌门中各有一个基因型的相似序列来自石油污染的西班牙 Cies群岛的潮下带沉积物,分别为基因型 C2_371(1个克隆子)和基因型 C2_329(1个克隆子),它们的相似序列分别为未培养酸杆菌克隆子OXIC-127和未培养拟杆菌克隆子RII-OX047。
虽然其它细菌类群在C2站点的系统发育树中所占比例较小,但典型基因型也有分布。硝化螺旋菌门中基因型 C2_13(2个克隆子)相似序列为东海大陆架表层沉积物的未培养硝化螺旋菌克隆子 DH132B19,绿弯菌门中基因型C2_314(2个克隆子)与富营养化湖泊沉积物中未培养绿弯菌克隆子 LakeCentre25有高同源性(99%),厚壁菌门中基因型C2_334(2个克隆子)与Bogota河中未培养厚壁菌克隆子RBE2CI-22有高同源性(99%)。
3 讨论
3.1 可培养细菌多样性
在对河口的微生物多样性研究中,多数采用未培养方法(姜丽晶,2007; 王新新,2008),但如果想对微生物的应用及防治有深入研究,可培养微生物依然至关重要(周丽华等,2009)。为研究椒江口沉积物中可培养细菌,本文使用了常规分类纯化、形态及生理生化指标和分子鉴定等方法,结果发现只有少数生理生化鉴定结果与分子鉴定结果一致,这一方面说明了生理生化鉴定由于其比对文库的局限性——生理生化鉴定的数据库主要针对医院以及疾控系统,海洋环境的菌株数据较少(王国良等,2008),因此很多生理生化鉴定和分子鉴定结果不能完全统一; 但另一方面,大多数菌株生理生化鉴定结果如果放在高的分类阶元(如门、纲、目等),其结果与分子鉴定结果将更加吻合,因此可以科学合理的利用生理生化与分子鉴定方法,加强可培养微生物的鉴定效率。

图8 C2站点根据细菌16S rDNA基因序列构建的系统发育树Fig.8 Phylogenetic tree of bacteria in 16S rDNA gene sequences in Station C2
近年来围绕海洋沉积物的可培养细菌多样性开展了较多研究,李昭(2013)对南太平洋环流区深海可培养细菌的多样性研究中发现,该区域的γ-变形菌纲在数量、种类、分布等方面都占主导地位(菌株数占81%); 卢婧雯等(2012)在对南海、东海近海可培养细菌的研究中发现,南海和东海分离菌株中γ-变形菌分别占35.5%和57.2%。上述研究从一定程度上证明γ-变形菌纲为沉积物中可培养细菌优势菌群,同时也有多个研究报道了厚壁菌门也是沉积物可培养细菌中的优势类群(Gontanget al,2007; 孙风芹等,2008;郑莹等,2012)。本实验对可培养细菌的研究结果(γ-变形菌纲与厚壁菌门占 63.8%和 31.7%)与霍颖异等(2008年)对浙江苍南近海沉积物可培养细菌多样性的研究结果(γ-变形菌纲与厚壁菌门占68.4%和29.1%)类似,也印证了上述结论。
值得注意的是,三个站点都有副溶血性弧菌(Vibrio parahaemolyticus)大量存在且分布均匀。副溶血性弧菌分为致病菌与非致病菌,其致病菌能引起食物中毒,主要分布在沿岸海水、海河交界处及海产食品中,有明显的季节性特点,夏秋季该菌量远大于其它季节(唐晓阳,2013)。本次采样时间为副溶血性弧菌丰度低的春季,其异常高的丰度提示该地区水产养殖业需要加强对副溶血性弧菌疫情的监控。
3.2 未培养细菌多样性
对于河口沉积物未培养细菌多样性的相关研究已有较多报道,姜丽晶(2007)对珠江口沉积物微生物多样性的研究表明: 变形菌门为最大优势菌群,其中δ-变形菌纲(主要为硫酸盐还原菌)为其主要组成部分,γ-变形菌纲则是第二优势菌群。王新新(2008)对长江口沉积环境细菌多样性的研究显示: δ-和γ-变形菌纲分别占变形菌门的53.4%和47.2%。Feng等(2009)对长江口及沿岸区域沉积物细菌多样性分析表明: γ-及δ-变形菌纲为该海域最主要的细菌类群。郭建丽等(2013)对双台子河口沉积物的研究结果显示,δ-变形菌纲为绝对优势类群,其次为γ-变形菌纲。上述研究都说明在河口环境中,变形菌门为最大优势类群,多数又以 δ-变形菌纲所占比例最高,其次为 γ-变形菌纲。但也有很多关于海洋沉积物的研究中,显示γ-变形菌纲才是未培养细菌的最主要类群(Bowmanet al,2003; Inagakiet al,2003; Websteret al,2006),王健鑫等(2012)对东海陆架表层沉积物的细菌多样性研究也发现: 变形菌门为最大优势菌群(占总克隆子数的41.5%),其中 γ-变形菌纲克隆子占比为 21.6%,这与本文对椒江口三个站点的研究结果(变形菌门和 γ-变形菌纲分别占比为42.7%和16.8%)相类似。椒江口相比长江和珠江河口,研究结果不尽相同,可能是由于椒江口区域的沉积物主要来自海域泥沙(谢钦春等,1998),而长江口及珠江口的沉积物主要来自河流流域(林承坤,1984; 江四义等,2008)。
Zhang等(2008)对维多利亚港及附近区域的沉积物的研究发现: 沉积物中的细菌类群与其所处环境是相互选择的,相似沉积环境中的细菌类群也类似。但本文中三个站点间的细菌类群存在很大差异,最为明显的是绿弯菌门,绿弯菌在 C0站点含量最高,占到26.55%,C2站点次之,占3.37%,C1站点则未扩增到绿弯菌16S rDNA序列,与此同时综合总碳、总氮、总磷等理化指标来看,站点 C0>C2>C1,因此猜测绿弯菌与沉积环境的理化指标结果有一定的关联性,也印证了绿弯菌门是水合物较少而有机质丰富的沉积物中的主要类群(李涛等,2008)。与绿弯菌相似,C1站点也没有检测到硝化螺旋菌的序列,硝化螺旋菌是一类好氧的硝化细菌,沉积物中的氧含量常能决定其分布(白洁等,2009),同时,它还对还原性硫化物敏感,一般存在于低硫环境(Kawaharaet al,2009),硝化螺旋菌的缺失可能暗示着C1站点沉积泥中溶解氧含量较低以及硫化物含量较高,这也与 C1站点沉积物中α-、δ-变形菌丰度高于另外两个站点相一致,因为这两类菌群在海洋沉积泥中主要是参与硫代谢(Jørgensen,1982; Nercessianet al,2005)。β-变形菌也是三个站点间有较明显差异的类群,一般在污染环境中 β-变形菌占较高比例(Brümmeret al,2003)。C1站点为含β-变形菌最多的站点,其中大多数克隆子的相似序列为陶厄氏菌(Thauera),而陶厄氏菌属的细菌具有相当广泛的污染物降解能力,即使较难降解的芳香族化合物,它们也普遍具有良好的降解力(毛跃建,2009)。Candidate division WS3是C2站点特有的类群,目前关于此类群的研究并不详细,但此类群主要出现在产甲烷环境(Dojkaet al,1998; Derakshaniet al,2001)。
3.3 环境与微生物的相关性
近些年相关研究显示,椒江口已经成为世界上已报道沉积物中含 PBDEs最高的区域之一,且春秋两季呈明显的富营养化状态(孙鲁峰等,2012; 杨华云等,2014)。赵永强等(2009)针对椒江口大型底栖动物群落结构的研究表明,由于受重金属和石油烃影响,耐污染能力较强的双鳃内卷齿蚕(Aglaophamusdibranchis)、光滑狭口螺(Stenothyra glabra)、日本大眼蟹(Macrophthalmus japonicus)、短拟沼螺(Assiminea brevicula)成为此地优势物种; 江锦花等在 2006和2007年对椒江口的鲻鱼(Mugil cephalus)和缢蛏(Sinonovacula constricta)的研究也表明,这两种典型生物富集了各种重金属及石油烃,且其富集量受沉积物及水体中污染物的浓度影响。
通过对系统发育树中相似序列来源环境的分析,可以更好地了解微生物群落与环境的关系(Clementinoet al,2007)。本实验研究发现,椒江口沉积物中许多典型细菌的相似序列都来自污染环境,如: 石油污染的西班牙 Cies群岛的潮下带沉积物(基因型C0_323、基因型C1_181)、原油污染的Berre潟湖沉积物(基因型 C0_129)、脂肪烃污染的土壤(基因型C0_6)、重金属污染的湘江沉积物(基因型 C1_133)、重金属污染的比利时大陆板块沉积物(基因型C2_337)、浙江东阳填埋场沉积物(基因型 C2_337)、高有机物输入的低氧缺氧淡水沉积物(基因型C1_113)、富营养化湖泊(基因型 C2_314)等环境,同时,还有一些相似序列为降解石油烃的可培养细菌(基因型C0_306、基因型C1_96)。综合其它研究者的结果,可以推测: 椒江口沉积物中的细菌多样性及群落结构已经受到重金属、石油烃以及水体富营养化等综合污染的深度影响。
4 结论
本实验通过常规分离纯化、鉴定和构建细菌克隆文库的方法,分别对椒江口三个站点沉积物中细菌的多样性及群落结构进行了研究,研究发现: (1) 对可培养细菌而言,分子鉴定方法较生理生化鉴定方法更加准确,γ-变形菌为三个站点的优势类群; (2) 对未培养方法得到的数据进行分析,C2站点多样性最为丰富,γ-变形菌为三个站点的最大优势类群; (3) 将系统发育树上的基因型与相似序列比较,相当数量的相似序列来自重金属或石油烃污染的沉积环境。
王国良,祝璨琳,金 珊,2008. 养殖大黄鱼(Pseudosciaena crocea)3种致病弧菌的分子鉴定及其系统发育学分析. 海洋与湖沼,39(2): 162—167
王金成,井明博,肖朝霞等,2012. 陇东黄土高原地区石油污染土壤微生物群落及其与环境因子的关系. 水土保持通报,32(5): 145—151
王健鑫,许贤恩,周链链等,2012. 东海陆架表层沉积物微生物多样性初步研究. 海洋与湖沼,43(4): 805—813
王新新,2008. 长江口、东海沉积环境细菌多样性和邻近西太平洋海域多环芳烃降解菌研究. 青岛: 中国海洋大学硕士学位论文,21—25
毛跃建,2009. 废水处理系统中重要功能类群 Thauera属种群结构与功能的研究. 上海: 上海交通大学博士学位论文,17—18
卢婧雯,张心齐,杜丽丽等,2012. 中国东海及南海近海 4采样点海水可培养细菌的多样性研究. 浙江大学学报(理学版),39(4): 443—449
白 洁,李海艳,赵阳国,2009. 黄海北部不同站位海洋细菌群落分布特征. 微生物学报,49(3): 343—350
齐海明,孙 岳,徐兆礼等,2013. 椒江口海域春秋季虾类群落结构及其影响因素. 应用生态学报,24(12): 3546—3552
江四义,郑兆勇,2008. 从珠江口沉积物粒度参数特征分析泥沙来源及其运移趋势. 中山大学学报(自然科学版),47(S1): 126—129
江锦花,朱利中,张明,2006. 椒江口水体和生物体中典型有机污染物的浓度水平及来源初探. 环境化学,25(5):546—549
江锦花,江正玲,陈希方等,2007. 椒江口海域重金属含量分布及在沉积物和生物体中的富集. 海洋环境科学,26(1):58—62
孙风芹,汪保江,李光玉等,2008. 南海南沙海域沉积物中可培养微生物及其多样性分析. 微生物学报,48(12):1578—1587
孙鲁峰,徐兆礼,邢小丽等,2012. 椒江口海域浮游植物与富营养化的关系. 应用与环境生物学报,18(2): 170—176
杜 萍,刘晶晶,沈李东等,2012. Biolog和PCR-DGGE技术解析椒江口沉积物微生物多样性. 环境科学学报,32(6):1436—1444
杜 萍,徐晓群,刘晶晶等,2011a. 椒江口春、秋季浮游动物分布特征及与主要环境因子的关系. 应用与环境生物学报,17(4): 486—494
杜 萍,刘晶晶,陈全震等,2011b. 椒江口春季水体异养细菌及氮、磷细菌的生态分布特征. 海洋环境科学,30(4):467—472
李 昭,2013. 南太平洋环流区深海可培养细菌的多样性研究以及两株海洋新菌的分类鉴定. 青岛: 中国海洋大学博士学位论文,61—63
李 涛,王 鹏,汪品先,2008. 南海西沙海槽表层沉积物微生物多样性. 生态学报,28(3): 1166—1173
李振高,骆永明,滕 应,2008. 土壤与环境微生物研究法.北京: 科学出版社,146—147
李海艳,2009. 黄海西北部真细菌群落结构及多样性研究. 青岛: 中国海洋大学硕士学位论文,38—47
杨华云,吕 伟,季天嘉等,2014. 椒江中下游沉积物中多溴联苯醚的分布特征. 杭州师范大学学报(自然科学版),13(2): 175—180
余 鹏,周珊珊,杨华云,2011. 椒江口沉积物中有机氯农药的含量及对映体特征. 环境科学与技术,34(11): 1—5
林承坤,1984. 长江口泥沙的来源分析与数量计算. 泥沙研究,(2): 22—32
周丽华,陈士超,邓志瑞等,2009. 太湖沉积物中的可培养细菌:Ⅰ. 细菌多样性初步分析. 湖泊科学,21(1): 27—35
郑 莹,霍颖异,2012. 浙江镇海潮间带沉积物中可培养微生物初步研究. 海洋学研究,30(4): 65—71
赵永强,陈全震,曾江宁等,2009. 椒江口化工园区及其邻近区域潮间带大型底栖动物秋冬季分布特征. 生态学报,29(3): 1578—1586
姜丽晶,2007. 珠江口沉积物微生物多样性及其与环境相互关系的研究. 厦门: 国家海洋局第三海洋研究所硕士学位论文,39—68
郭建丽,樊景凤,付 慧等,2013. 双台子河口沉积物中细菌多样性分析. 微生物学通报,40(9): 1550—1559
唐晓阳,2013. 水产品中副溶血性弧菌风险评估基础研究. 上海: 上海海洋大学博士学位论文,2—3
黄 备,闵 怀,唐静亮等,2009. 浙江沿海海洋微生物群落的研究. 中国环境监测,25(2): 44—47
谢钦春,李伯根,1998. 椒江河口悬沙浓度垂向分布和泥跃层发育. 海洋学报,20(6): 58—59
霍颖异,许学伟,王春生等,2008. 浙江苍南近海沉积物细菌物种多样性. 生态学报,28(10): 5166—5172
Magurran A E编著,张 峰译,2011. 生物多样性测度. 北京:科学出版社,66—67
Bowman J P,McCuaig R D,2003. Biodiversity,community structural shifts,and biogeography of prokaryotes within Antarctic continental shelf sediment. Applied and Environmental Microbiology,69(5): 2463—2483
Brümmer I H M,Felske A,Wagner-Döbler I,2003. Diversity and seasonal variability of β-proteobacteria in biofilms of polluted rivers: analysis by temperature gradient gel electrophoresis and cloning. Applied and Environmental Microbiology,69(8): 4463—4473
Clementino M M,Fernandes C C,Vieira R Pet al,2007.Archaeal diversity in naturally occurring and impacted environments from a tropical region. Journal of Applied Microbiology,103(1): 141—151
Derakshani M,Lukow T,Liesack W,2001. Novel bacterial lineages at the (Sub) division level as detected by signature nucleotide-targeted recovery of 16S rRNA genes from bulk soil and rice roots of flooded rice microcosms. Applied and Environmental Microbiology,67(2): 623—631
Dojka M A,Hugenholtz P,Haack S Ket al,1998. Microbial diversity in a hydrocarbon-and chlorinated- solvent-contaminated aquifer undergoing intrinsic bioremediation. Applied and Environmental Microbiology,64(10): 3869—3877
Feng B W,Li X R,Wang J Het al,2009. Bacterial diversity of water and sediment in the Changjiang estuary and coastal area of the East China Sea. FEMS Microbiology Ecology,70(2): 236—248
Gontang E A,Fenical W,Jensen P R,2007. Phylogenetic diversity of gram-positive bacteria cultured from marine sediments. Applied and Environmental Microbiology,73(10): 3272—3282
Heijs S K,Laverman A M,Forney L Jet al,2008. Comparison of deep-sea sediment microbial communities in the Eastern Mediterranean. FEMS Microbiology Ecology,64(3):362—377
Hu B,Shen L,Du Pet al,2012. The influence of intense chemical pollution on the community composition,diversity and abundance of anammox bacteria in the Jiaojiang Estuary(China). PLoS One,7(3): e33826
Inagaki F,Suzuki M,Takai Ket al,2003. Microbial communities associated with geological horizons in coastal subseafloor sediments from the Sea of Okhotsk. Applied and Environmental Microbiology,69(12): 7224—7235
Jørgensen B B,1982. Mineralization of organic matter in the sea bed-the role of sulphate reduction. Nature,296(5858): 643—645
Kawahara N,Shigematsu K,Miyadai Tet al,2009. Comparison of bacterial communities in fish farm sediments along an organic enrichment gradient. Aquaculture,287(1—2):107—113
Kumar S,Tamura K,Nei M,2004. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Briefings in Bioinformatics,5(2): 150—163
Li T,Wang P,Wang P A,2008. Microbial diversity in surface sediments of the Xisha Trough,the South China Sea. Acta Ecologica Scinica,28(3): 1166—1173
Nercessian O,Fouquet Y,Pierre Cet al,2005. Diversity of Bacteria and Archaea associated with a carbonate-rich metalliferous sediment sample from the Rainbow vent field on the Mid-Atlantic Ridge. Environmental Microbiology,7(5): 698—714
Nogales B,Moore E R B,Llobet-Brossa Eet al,2001. Combined use of 16S ribosomal DNA and 16S rRNA to study the bacterial community of polychlorinated biphenyl-polluted soil. Applied and Environmental Microbiology,67(4): 1874—1884
Schloss P D,Handelsman J,2005. Introducing DOTUR,a computer program for defining operational taxonomic units and estimating species richness. Applied and Environmental Microbiology,71(3): 1501—1506
Shen L D,Zhu Q,Liu Set al,2014. Molecular evidence for nitrite-dependent anaerobic methane-oxidising bacteria in the Jiaojiang Estuary of the East Sea (China). Applied Microbiology and Biotechnology,98(11): 5029—5038
Webster G,Parkes R J,Cragg B Aet al,2006. Prokaryotic community composition and biogeochemical processes in deep subseafloor sediments from the Peru Margin. FEMS Microbiology Ecology,58(1): 65—85
Yue X,Liu B Z,Xiang J Het al,2010. Identification and characterization of the pathogenic effect of aVibrio parahaemolyticus-related bacterium isolated from clamMeretrix meretrixwith mass mortality. Journal of Invertebrate Pathology,103(2): 109—115
Zhang W,Ki J-S,Qian P Y,2008. Microbial diversity in polluted harbor sediments I: bacterial community assessment based on four clone libraries of 16S rDNA. Estuarine,Coastal and Shelf Science,76(3): 668—681
Zhu J Y,Zhang J X,Li Qet al,2013. Phylogenetic analysis of bacterial community composition in sediment contaminated with multiple heavy metals from the Xiangjiang River in China. Marine Pollution Bulletin,70(1—2): 134—139
猜你喜欢
东北亚学刊(2022年3期)2022-02-04
中国比较医学杂志(2020年4期)2020-05-26
江苏农业科学(2019年5期)2019-09-02
浙江警察学院学报(2019年4期)2019-09-02
水生生物学报(2019年4期)2019-07-20
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
广东农业科学(2017年5期)2017-08-29
传媒评论(2017年2期)2017-06-01
浙江柑橘(2016年3期)2016-03-11
