
碘造影剂诱导糖尿病大鼠肾细胞凋亡涉及Akt/mTOR信号途径探讨
2015-03-06 08:38焦占全陈军刘艳红李广平
中国心血管杂志 2015年3期
焦占全 陈军 刘艳红 李广平
·基础研究·
碘造影剂诱导糖尿病大鼠肾细胞凋亡涉及Akt/mTOR信号途径探讨
焦占全 陈军 刘艳红 李广平
目的 探讨肾细胞凋亡在糖尿病大鼠造影剂急性肾损害(CIAKI)发病中的作用,并研究其对Akt/mTOR信号途径的影响。方法 雄性SD大鼠24只,遵循体质量分层随机原则分为正常对照组(N组)、正常+造影剂组(NC组)、糖尿病对照组(D组)和糖尿病+造影剂组(DC组),每组6只。以腹腔单剂量注射链脲佐菌素建立糖尿病模型;10周后经股静脉注射60%泛影葡胺(10 ml/kg),连续2 d,建立CIAKI模型24 h后处死大鼠留取标本,测血、尿肌酐值;免疫组化法检测肾内凋亡相关蛋白caspase-3的表达;Western Blot定量检测肾组织内Bcl-2、Bax及Akt/mTOR信号通路关键蛋白的表达。结果 与D组比较,DC组注射泛影葡胺后血肌酐显著增加[(103.89±9.01)μmol/L比(71.52±7.03)μmol/L,P=0.000],而肌酐清除率显著降低[(1.49±0.33)ml/min比(2.60±0.54)ml/min,P=0.001];肾内凋亡相关蛋白caspase-3阳性表达量显著增加(17.55±0.86比10.02±1.48,P=0.000);抗凋亡Bcl-2蛋白表达则显著下降,而促凋亡Bax蛋白表达显著增加(0.90±0.13比 1.50±0.16;0.92±0.04比 0.51±0.05,均为P=0.000)。糖尿病CIAKI时上游p-Akt和p-mTOR均表达下调(0.46±0.08比0.68±0.07,P=0.002;0.19±0.04比0.50±0.07,P=0.000)。结论 离子型高渗造影剂可能通过抑制糖尿病大鼠Akt/mTOR信号通路磷酸化激活介导肾细胞经线粒体caspase-3途径凋亡而导致急性肾损伤。
糖尿病; 造影剂急性肾损害; 细胞凋亡; Akt/mTOR信号通路
造影剂急性肾损害(contrast induced acute kidney injury,CIAKI)已成为医院内获得性急性肾功能衰竭(acute renal failure,ARF)的主要原因之一。而糖尿病肾病(diabetic nephropathy,DN)是发生CIAKI最常见的危险因素[1-2]。细胞研究发现,无论是高渗还是低渗造影剂,在高血糖(30 mmol/L)条件下细胞凋亡均显著加重[3-4],因此造影剂与高血糖可能对细胞凋亡有协同作用。哺乳动物西罗莫司靶蛋白(mammalian target of rapamycin,mTOR)是一个进化上高度保守的丝氨酸/苏氨酸蛋白激酶,处于生物体内复杂信号网络的中心环节,是体内上游信号的信号整合分子[5-6]。而Akt是细胞生存的关键调节者,mTOR是其下游效应信号之一,Akt/mTOR信号传导通路的活化可促进细胞周期进展,从而促进细胞的生存和增殖。Rane等[7]发现,体外培养的肾小管细胞在持续高糖环境下出现细胞凋亡,其机制与Akt活化的抑制有关,但目前有关糖尿病时造影剂诱导肾细胞凋亡机制的体内实验研究尚未见报道。因此,本研究拟建立可靠的糖尿病CIAKI模型,在体内实验条件下探讨肾细胞凋亡在糖尿病大鼠CIAKI发病中的作用,并研究其对Akt/mTOR信号途径的影响。
1 材料与方法
1.1 实验动物和主要试剂
健康雄性清洁级SD大鼠24只,8周龄,体重(250±20)g,购自中国人民解放军军事医学科学院实验动物中心。链脲佐菌素购自Sigma公司;60%泛影葡胺购自上海旭东海普药业有限公司;血清肌酐(Scr)试剂盒购自中生北控生物科技股份有限公司;caspase-3兔抗大鼠多克隆抗体购自武汉博士德生物工程有限公司;Bcl-2抗体、Bax抗体、Phospho-Akt(Ser473)抗体和Phospho-mTOR(Ser2448)抗体购自Cell Signaling公司。
1.2 糖尿病大鼠CIAKI模型的建立与分组
给予实验动物标准鼠食,自由摄食水,遵循体重分层随机原则分为两组:正常组(12只),糖尿病组(12只)。禁食12 h,糖尿病组大鼠以腹腔单剂量注射链脲佐菌素60 mg/kg建立糖尿病模型,注射后72 h及1周断尾取血测血糖,以非禁食血糖≥16.7 mmol/L为标准判定糖尿病大鼠模型是否成功。实验期间每周测血糖1次,保证血糖≥16.7 mmol/L。
将正常组大鼠再遵循体重分层随机原则分为正常对照组(N组,n=6)和正常+造影剂组(NC组,n=6);将糖尿病组大鼠再遵循体重分层随机原则分为糖尿病对照组(D组,n=6)和糖尿病+造影剂组(DC组,n=6)。此后继续给予普通饲料,自由摄水,饲养满10周后NC组和DC组以10%水合氯醛(3 ml/kg)经腹腔注射麻醉大鼠,采用小切口逐层切开,分离出左(右)股静脉,经静脉留置针注射60%泛影葡胺(10 ml/kg,推注时间不少于10 min),1次/d连续2 d,建立CIAKI模型;N组和D组注射同等量生理盐水。
1.3 标本采集
造模结束后将大鼠放入代谢笼,留取24 h尿液并记录尿量。然后以10%水合氯醛(3 ml/kg)经腹腔注射麻醉大鼠,经腹部正中切口,经下腔静脉抽血5 ml留验肾功能等生化指标。经腹主动脉用冰冻生理盐水充分灌洗至流出液体清亮。分离两肾包膜,迅速取下双肾。左肾去包膜后用10%中性甲醛固定,石蜡包埋后切片行相关染色;右肾分离后迅速保转至-80℃冰箱冻存,用于Western Blot蛋白定量分析。
1.4 血、尿生化指标检测
按照试剂盒说明书,利用半自动生化仪测定Scr及尿肌酐水平,并计算肌酐清除率。计算公式:肌酐清除率=(尿肌酐×24 h尿量)/(Scr×24×60×体重)。
1.5 免疫组化检测肾组织凋亡相关蛋白caspase-3表达
石蜡切片常规脱蜡、水化,微波抗原修复,室温孵育15 min,消除内源性过氧化物酶,滴加caspase-3兔抗大鼠多克隆抗体,再滴加相应的生物素标记的二抗,以磷酸盐缓冲液代替一抗作为阴性对照,加底物3,3′-二甲基联苯氨显色,阳性表达呈棕黄色着色,光镜下观察。每张切片随机测定10个完整而不重叠视野,在校正光密度后测定每个视野下阳性反应的累积光密度(optical density,IOD),以累积光密度代表含量密度。
1.6 Western Blot定量分析凋亡调节蛋白Bcl-2、Bax及Akt/mTOR信号通路蛋白的表达
组织块称重、粉碎,裂解液裂解,考马斯亮蓝法测定蛋白浓度。上样、10% SDS-PAGE电泳后,将蛋白转印到PVDF膜上。用5%脱脂奶粉37℃封闭2 h;加相应一抗4℃过夜,加辣根过氧化酶标记的二抗室温孵育2 h,用ECL化学发光法检测,X线片显影成像,将显色条带扫描至计算机中,使用Image J分析软件分析底片上条带的光密度值,以靶蛋白/内参GAPDH光密度比值作为目标蛋白的相对表达丰度。
1.7 统计学方法
2 结果
2.1 糖尿病大鼠CIAKI模型的成功建立
糖尿病大鼠Scr水平与正常大鼠相似,而肌酐清除率稍高(P>0.05);NC组大鼠注射造影剂后Scr水平稍高于N组,而肌酐清除率水平略低于N组,但差异均无统计学意义(均为P>0.05)。值得注意的是,DC组大鼠注射高渗造影剂后Scr水平显著高于D组[(103.89±9.01)μmol/L比(71.52±7.03)μmol/L,P<0.05],肌酐清除率显著低于D组[(1.49±0.33)ml/min比(2.60±0.54)ml/min,P<0.05],见表1。
2.2 免疫组化法比较各组凋亡相关蛋白caspase-3阳性表达量(×105)
NC组大鼠肾脏caspase-3阳性表达量较N组略升高,但差异无统计学意义(5.48±0.36比4.71±0.28,P=0.151)。D组大鼠肾脏caspase-3阳性表达量较N组明显升高(10.02±1.48比4.71±0.28,P=0.000);而DC组caspase-3阳性表达量显著高于D组(17.55±0.86比10.02±1.48,P=0.000),见图1。
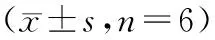
表1 各组大鼠血肌酐和肌酐清除率比较
注:与N组比较,aP<0.05;与NC组比较,bP<0.05;与D组比较,cP<0.05
A:N组,正常对照组;B:NC组,正常+造影剂组;C:D组,糖尿病对照组;D:DC组,糖尿病+造影剂组
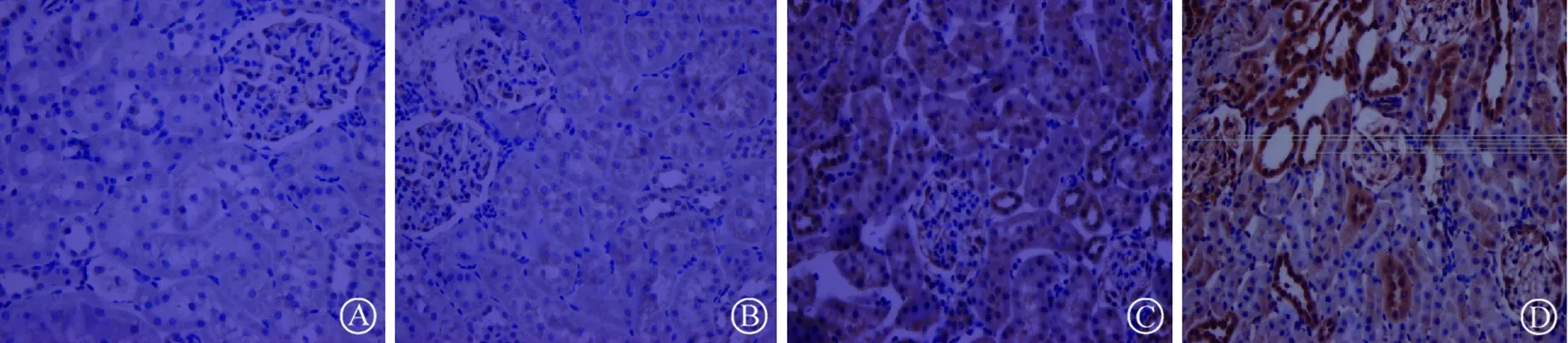
2.3 线粒体Bcl-2家族Bcl-2、Bax蛋白表达的变化
N:正常对照组;NC:正常+造影剂组;D:糖尿病对照组;DC:糖尿病+造影剂组
与N组相比,D组大鼠肾脏Bcl-2蛋白表达显著降低(1.50±0.16比2.56±0.47,P=0.008);NC组较N组略有下降,但两组比较差异无统计学意义(2.24±0.37比2.56±0.47,P=0.720)。DC组大鼠肾脏Bcl-2的表达较D组显著下降(0.90±0.13比1.50±0.16,P=0.000)。而Bax蛋白表达趋势与之相反,DC组大鼠肾脏Bax蛋白表达较D组显著增加(0.92±0.04比0.51±0.05,P=0.000),见图2。
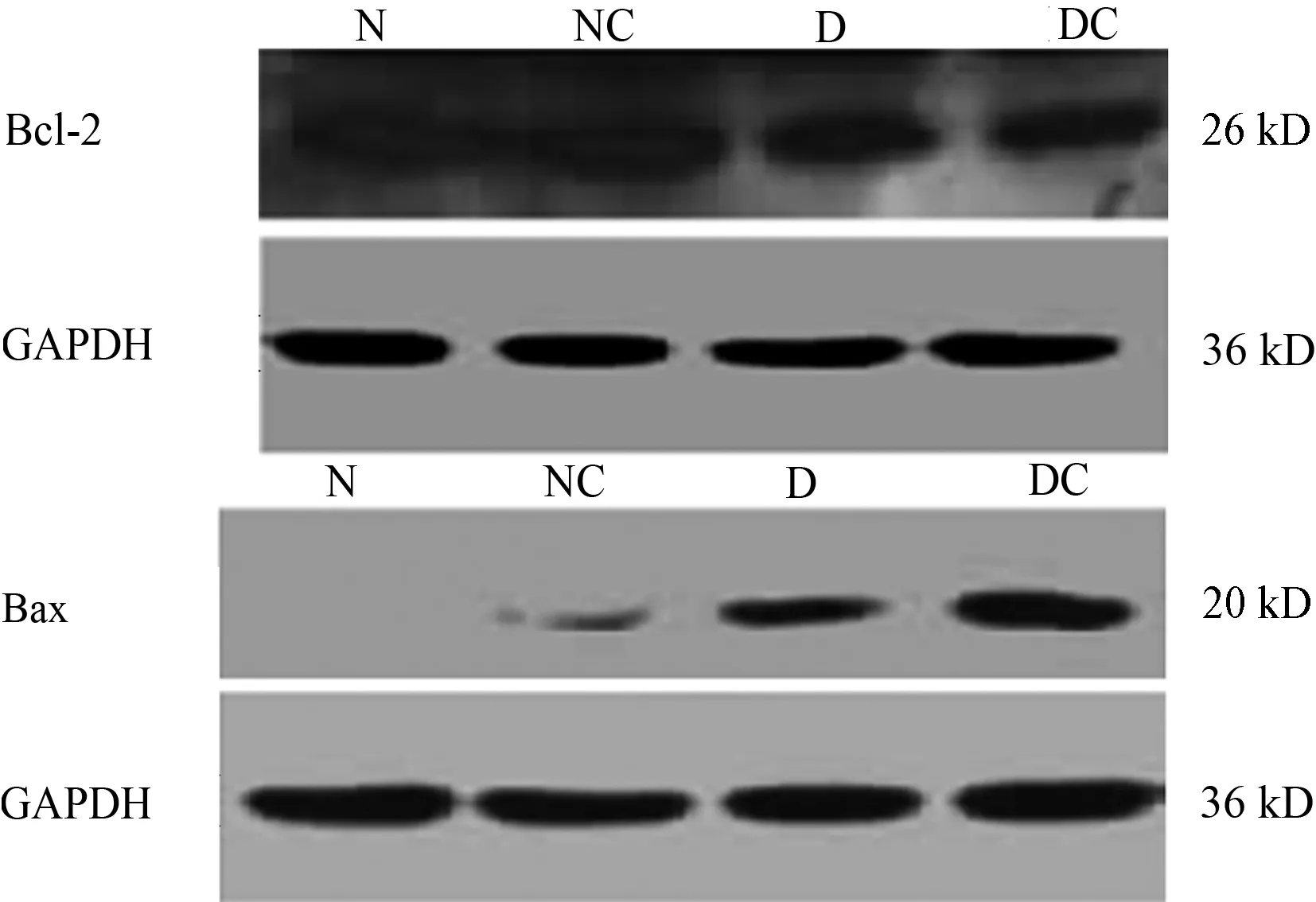
2.4 CIAKI时Akt/mTOR信号途径蛋白表达变化
NC组、D组大鼠肾脏p-Akt(S473)的表达显著低于N组(0.72±0.11比1.01±0.14; 0.68±0.07比1.01±0.14,均P=0.000);DC组大鼠肾脏p-Akt(S473)的表达较D组显著下降(0.46±0.08比0.68±0.07,P=0.002)。与之相似,p-Akt(S473)的下游信号mTOR其磷酸化激活程度,NC组、D组均显著低于N组(0.73±0.14比0.95±0.12,P=0.001;0.50±0.07比 0.95±0.12,P=0.000);DC组大鼠肾脏p-mTOR的表达较D组显著下降(0.19±0.04比0.50±0.07,P=0.000),见图3。
N:正常对照组;NC:正常+造影剂组;D:糖尿病对照组;DC:糖尿病+造影剂组
3 讨论
目前,糖尿病是全球第3大非传染性“流行性”疾病,其代表发生CIAKI和CIAKI相关心血管死亡的高危风险[1-2]。故此,我们确定了本研究诱导早期糖尿病肾病,并在此基础上注射高渗造影剂泛影葡胺以诱导建立糖尿病CIAKI模型的方案。最大限度地模拟CIAKI临床实际进程,以使实验得出的结论更具代表性和说服力。
本实验数据表明,与正常大鼠相比,病程10周时糖尿病大鼠虽然Scr无明显升高,但肌酐清除率近乎达到正常大鼠的两倍,表明其处于早期糖尿病肾病阶段。如前所述,正常大鼠对CIAKI的发生有“抵抗力”,而糖尿病大鼠注射泛影葡胺后肌酐清除率显著下降,这表明大鼠在糖尿病肾病状态下对于离子型高渗造影剂泛影葡胺更为敏感,肾功能损伤程度更加严重。
近年研究发现,高血糖状态下肾小管上皮细胞凋亡增加,推测细胞凋亡参与了DN的发生[8-9]。进一步的体外研究也证实,造影剂对高糖状态下的肾细胞凋亡有显著影响。Wasaki等[3]研究发现,高糖条件增强了肾小球系膜细胞对造影剂细胞毒性的敏感性。O′Donnell等[4]体外培养MDCK细胞,发现无论是高渗还是低渗造影剂,在高血糖(30 mmol/L)条件下细胞凋亡均显著加重,故而他们提出造影剂与高血糖对细胞凋亡有协同作用。而目前有关糖尿病时造影剂诱导肾细胞凋亡的体内研究尚未见报道,本研究在体内实验条件下证实糖尿病大鼠应用高渗造影剂后肾细胞凋亡相关蛋白caspase-3表达显著升高,这表明离子型高渗造影剂会加重糖尿病大鼠肾细胞凋亡。许多体外研究已证实,造影剂可影响Bcl-2基因家族蛋白的表达而诱导肾细胞凋亡[10-15]。Yano等[12]研究发现,造影剂诱导LLC-PK1细胞凋亡,这一损伤或许是因为下调了Bax/Bcl-2表达,随后增加了caspase-9和caspase-3活性。同样,我们在体内实验也证实机体在糖尿病基础状态下,离子型高渗造影剂可能通过某种相应特定的信号转导打破了Bcl-2基因家族中Bcl-2与Bax之间的“平衡”,Bcl-2/Bax比值下调,此后完全启动线粒体caspase-3信号通路,促进肾细胞凋亡。
如前所述,Akt/mTOR信号传导通路的活化可促进翻译过程的启动,促进RNA和蛋白质合成,从而促进细胞的生存和增殖。Brognard等[16]发现,天然肿瘤抑制蛋白PHLPP可使Akt去磷酸化而失活,从而抑制细胞周期,促进细胞凋亡。亦有研究发现,mTORC1的活化能对抗UV诱导的细胞凋亡。Faghiri和Bazan[17]研究PI3K/Akt和mTOR/P70S6K途径可传递视网膜色素上皮细胞遭受氧化应激时的部分生存信号。近来,Rane 等[7]发现体外培养的肾小管细胞在持续高糖环境下出现细胞凋亡,其机制与Akt活化的抑制有关。Andreucci等[18]通过体外研究发现,Akt/mTOR/P70S6K途径去磷酸化失活参与了造影剂引起的肾小管上皮细胞凋亡。本研究中各组大鼠肾脏p-Akt(S473)及其下游信号分子p-mTOR的表达趋势基本一致:D组低于N组;DC组较D组显著下降。表明与正常大鼠相比,早期糖尿病肾病阶段Akt/mTOR信号途径磷酸化激活受到抑制;更值得关注的是离子型高渗造影剂诱导糖尿病大鼠肾脏Akt/mTOR途径去磷酸化失活进一步加重。这也验证了以往体外实验的结果,进一步证实Akt/mTOR信号途径参与抗凋亡机制,泛影葡胺可经Akt/mTOR途径去磷酸化失活而加重糖尿病大鼠肾细胞凋亡。
此外,我们既往的研究还发现,已存在基础肾损伤时注射造影剂更易导致肾脏氧化应激状态改变,诱发急性肾损伤[19]。糖尿病大鼠注射高渗造影剂泛影葡胺后触发肾内一系列的信号途径的表达改变,上游Akt/mTOR信号途径显著去磷酸化失活,机体细胞抗凋亡防御能力下降,这些上游信号的整合结果影响了Bcl-2/Bax比值,经过内源性线粒体caspase-3途径诱导肾细胞凋亡。可见糖尿病时高渗造影剂促发的肾细胞凋亡是上述信号“网络”的共同调节传递信息的结果。总之,离子型高渗造影剂可能通过抑制糖尿病大鼠Akt/mTOR信号通路磷酸化激活介导肾细胞经线粒体caspase-3途径凋亡而导致急性肾损伤。
[1] Waybill MM, Waybill PN. Contrast-media induced nephrotoxicity: identification of patients at risk and algorithms for prevention[J]. J Vasc Interv Radiol, 2001, 12: 3-9.
[2] Pflueger AC, Larson TS, Nath KA, et al. Role of adenosine in contrast media-induced acute renal failure in diabetes mellitus[J]. Mayo Clin Proc, 2000, 75: 1275-1283.
[3] Wasaki M, Sugimoto J, Shirota K. Glucose alters the susceptibility of mesangial cells to contrast media[J]. Invest Radiol, 2001, 36: 355-362.
[4] O′Donnell DH, Moloney MA, Bouchier-Hayes DJ. Contrast-induced nephro- toxicity: possible synergistic effect of stress hyperglycemia[J]. AJR Am J Roentgenol, 2010, 195: W45-W49.
[5] Inoki K, Zhu T, Guan KL, et al. TSC2 mediates cellular energy response to control cell growth and survival[J]. Cell, 2003, 115: 577-590.
[6] Inoki K, Ouyang H, Li Y, et al. Signaling by target of rapamycin proteins in cell growth control[J]. Microbiol Mol Biol Rev, 2005, 69: 79-100.
[7] Rane MJ, Song Y, Jin S, et al. Interplay between Akt and p38 MAPK pathways in the regulation of renal tubular cell apoptosis associated with diabetic nephropathy[J]. Am J Physiol Renal Physiol, 2009, 298: F49-F61.
[8] Nakagami H, Morishita R, Yamamoto K, et al. Hepatocyte growth factor prevents endothelial cell death through inhibition of bax translocation from cytosol to mitochondria membrane[J]. Diabetes, 2002, 51: 2604-2611.
[9] Ma YX, Bai GH, Bai XY, et al. The mechanism of apoptosis induced by high glucose in human renal tubular epithelial cells[J]. J Qinghai Med Coll, 2009, 3: 150-154. (in Chinese) 马玉香, 白光辉, 白雪源, 等. 高糖诱导人肾小管上皮细胞凋亡的机制研究[J]. 青海医学院学报, 2009, 3: 150-154.
[10] Romano G, Briguori C, Quintavalle C, et al. Contrast agents and renal cell apoptosis[J]. Eur Heart J, 2008, 29: 2569-2576.
[11] Yano T, Itoh Y, Kubota T, et al. A prostacyclin analog prevents radiocontrast nephropathy via phosphorylation of cyclic AMP response element binding protein[J]. Am J Pathol, 2005, 166: 1333-1342.
[12] Yano T, Itoh Y, Sendo T, et al. Cyclic AMP reverses radiocontrast media-induced apoptosis in LLC-PK1 cells by activating A kinase/PI3 kinase[J]. Kidney Int, 2003, 64: 2052-2063.
[13] Xiong XL, Jia RH, Yang DP, et al. Irbesartan attenuates contrast media-induced apoptosis of NRK-52E cells[J]. Chin J Nephrol, 2006, 22: 682-687. (in Chinese) 熊晓玲, 贾汝汉, 杨定平, 等. 依贝沙坦抑制造影剂诱导的肾小管上皮细胞凋亡[J]. 中华肾脏病杂志, 2006, 22: 682-687.
[14] Lee HC, Sheu SH, Yen HW, et al. JNK/ATF2 pathway is involved in iodinated contrast media-induced apoptosis[J]. Am J Nephrol, 2010, 31: 125-133.
[15] Kolyada AY, Liangos O, Madias NE, et al. Protective effect of erythropoietin against radiocontrast-induced renal tubular epithelial cell injury[J]. Am J Nephrol, 2008, 28: 203-209.
[16] Brognard J, Sierecki E, Gao T, et al. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms[J]. Mol Cell, 2007, 25: 917-931.
[17] Faghiri Z, Bazan NG. PI3K/Akt and mTOR/p70S6K pathways mediate neuroprotectin D1-induced retinal pigment epithelial cell survival during oxidative stress-induced apoptosis[J]. Exp Eye Res, 2010, 90: 718-725.
[18] Andreucci M, Fuiano G, Presta P, et al. Radiocontrast media cause dephosphorylation of Akt and downstream signaling targets in human renal proximal tubular cells[J]. Biochem Pharmacol, 2006, 72: 1334-1342.
[19] Chen J, Jiao ZQ, Li GP, et al. Relationship between the expression of myeloperoxidase and nitric oxide in contras-induced nephropathy and oxidative stress[J]. Chin Foreign Med Res, 2012, 10: 1-3. (in Chinese) 陈军, 焦占全, 李广平,等. MPO、NO在造影剂肾病中的表达及其与氧化应激的关系[J].中外医学研究,2012,10:1-3.
(本文编辑:谭潇)
Akt/mTOR expression is involved in iodinated contrast media induced renal cell apoptosis in diabetic rats
JiaoZhanquan1,ChenJun1,LiuYanhong2,LiGuangping1.
1TianjinKeyLaboratoryofIonic-MolecularFunctionofCardiovascularDisease,TianjinInstituteofCardiology,DepartmentofCardiology,theSecondHospitalofTianjinMedicalUniversity,Tianjin300211,China; 2DepartmentofCardiology,TianjinThirdCentralHospital
LiGuangping,Email:tjcardiol@126.com
Objective To investigate molecular mechanisms of renal cell apoptosis on CIAKI in vivo, especially the involvement of Akt/mTOR signal pathways. Methods Diabetic Sprague-Dawley rats were induced by intraperitoneal injection of streptozotocin. Ten weeks later the normal and diabetic rats were administered 60% meglumine diatrizoate (DTZ) or normal saline (10 ml/kg) injection for two days continuously. 24 h after the operation, the rats were killed, stored blood samples for examining blood creatinine and kidneys for immunohistochemistry and western blotting analysis. The expression of caspase-3 in the kidney was investigated by immunohistochemistry. Meanwhile, quantitative analysis of the Bcl-2, Bax, upstream signal molecule p-Akt and p-mTOR protein expression by western blotting was used to study renal cell apoptosis on CIAKI. Results Compared with diabetic group (D group), the serum creatinine was significantly increased in diabetes+contrast media (DTZ) group (DC group) after operation [(103.89±9.01)μmol/Lvs. (71.52±7.03)μmol/L,P=0.000]. The creatinine clearance rate (Ccr) was also significantly decreased in DC group after operation[(1.49±0.33)ml/minvs. (2.60±0.54)ml/min,P=0.001]. Especially in the diabetic kidney, the expression of caspase-3 was also significantly increased after intravenous injection HOCM compared with normal saline. And the expression of anti-apoptosis Bcl-2 protein was significantly decreased in DC group after the induction of DTZ, while the expression of promoting apoptosis protein Bax was significantly increased (0.90±0.13vs. 1.50±0.16; 0.92±0.04vs. 0.51±0.05, bothP=0.000). The activity of upstream signal molecule p-Akt and p-mTOR was significantly decreased (0.46±0.08vs. 0.68±0.07,P=0.002; 0.19±0.04vs. 0.50±0.07,P=0.000). Conclusions The ionic high osmolality CM induce severe renal cells apoptosis in diabetic rats through activating the caspase-3 apoptotic pathway that may be mediated by upstream Akt/mTOR (inhibiting p-Akt and p-mTOR expression) signal pathways.
Diabetes mellitus; Contrast induced acute kidney injury; Apoptosis; Akt/mTOR signal pathways
10.3969/j.issn.1007-5410.2015.03.012
天津市卫生局科技基金资助项目(2012KZ079)
300211 天津市心血管病离子与分子机能重点实验室 天津医科大学第二医院心脏科 天津心脏病学研究所(焦占全、陈军、李广平);天津市第三中心医院心脏科(刘艳红)
李广平,电子邮箱:tjcardiol@126.com
2014-10-21)
ThisworkwassupportedbyagrantfromtheScienceandTechnologyFoundationofTianjinMunicipalHealthBureau(No.2012KZ079)
猜你喜欢
新世纪智能(数学备考)(2021年10期)2021-12-21
现代仪器与医疗(2021年4期)2021-11-05
新世纪智能(数学备考)(2020年10期)2021-01-04
中华养生保健(2020年4期)2020-11-16
家庭医药(2018年9期)2018-09-27
中成药(2017年12期)2018-01-19
祝您健康(2017年6期)2017-06-06
中国交通信息化(2017年8期)2017-06-06
家庭医药(2016年11期)2016-11-24
现代养生·上半月(2016年8期)2016-05-14
