
GCLE 的合成路线研究
2015-02-20 12:04孙鹏
当代化工研究 2015年2期
孙鹏
(菏泽市机电石化协会 山东菏泽 274000)
一、 简介
GCLE 化学名为7- 苯乙酰胺-3- 氯甲基头孢烷烯酸对甲氧基苄酯,英文名称为4-methoxybezyl-3-(chloromethyl)-8-oxo-7-(2-phenylacetamido)-5-t h i a-l-a z a b i c y c l o【4.2.0】o c t-e n e-2-carboxylate, 结构式为:
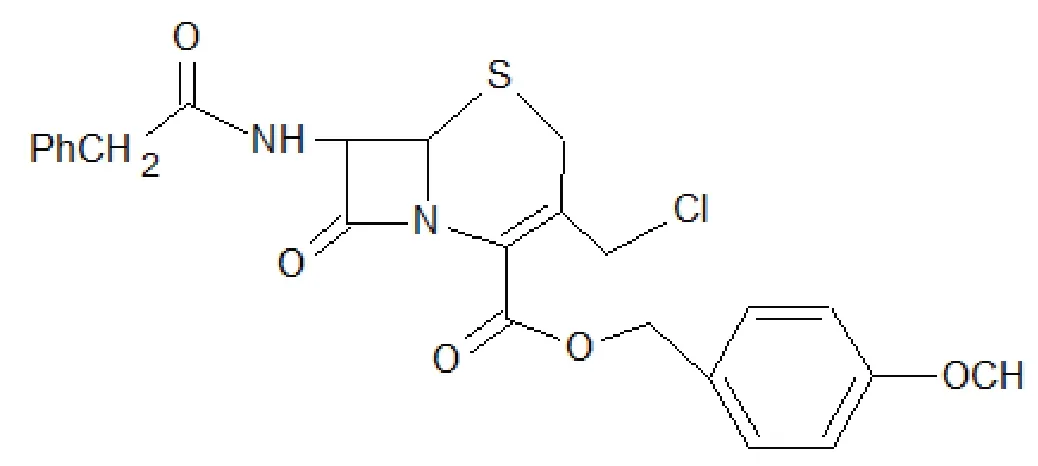
GCLE 是继7-ACA 和7-ADCA 后合成头孢菌素类药物的又一重要中间体(三者被称为头孢菌素三大母核)。以7-ACA 为中间体修饰C3- 及C4- 位侧链进行各种头孢菌素的合成,因其价格昂贵,限制了以其为中间体的头孢菌素的生产。由于7-ADCA的C3- 位的甲基难再进行化学修饰,限制了功能团的引入,而GCLE 的C3- 位氯甲基基团具有化学活泼性,活化了许多C3- 位的取代反应,从该关键中间体可以在温和的反应条件下方便地制取各种类型的新半合成头孢菌素。和前两者相比,GCLE 不仅实现了C3- 位的功能改造,而且是以廉价的青霉素为原料,通过化学转化而得到的抗菌谱广、对β- 内酰胺酶更稳定的头孢中间体,因此积极开发GCLE 有重要意义。
二、 合成路线研究
目前合成GCLE 的路线较多,但主要分为两类6 种。这两类是扩环后卤代与扩环前卤代,每类又分为3 种路线。
第一类:扩环后卤代
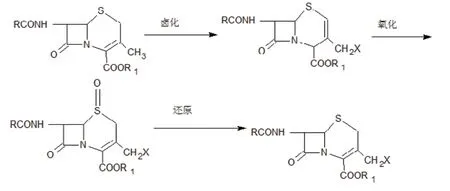
1、直接卤化。3- 环外亚甲基头孢衍生物主要早期用作头孢克洛的关键中间体。Morin 等在青霉素氧化物扩环制备去氧乙酰基头孢时,分离出少量的- 环外亚甲基头孢中间体。1976 年,Kukolja 等发现了将青霉素钾盐经过氧化、酯化保护、氯代开环和闭环等步骤合成3- 环外亚甲基头孢衍生物,该法成为青霉素扩环制备3- 环外亚甲基化合物主要方法。早期的Kukol ja 方法产率较低,后人又对其方法进行了改进。Koppel 发现3- 环外亚甲基头孢中间体可又直接卤化,得到3- 卤甲基头孢菌素衍生物,因此开辟了一条用3- 环外亚甲基头孢衍生物合成GCLE 的方法,合成路线如下:

2、光溴化。关于以7-ADCA 为原料通过光溴化法制备3- 卤代甲基化合物的报道较前就有, 由3-去乙酰氧基头孢直接进行烯丙位溴化,卤化后容易形成△2- 烯而失去药效,因此必须将△2- 烯氧化为△3- 烯,再还原为3- 卤代甲基头孢,反应路线如下:
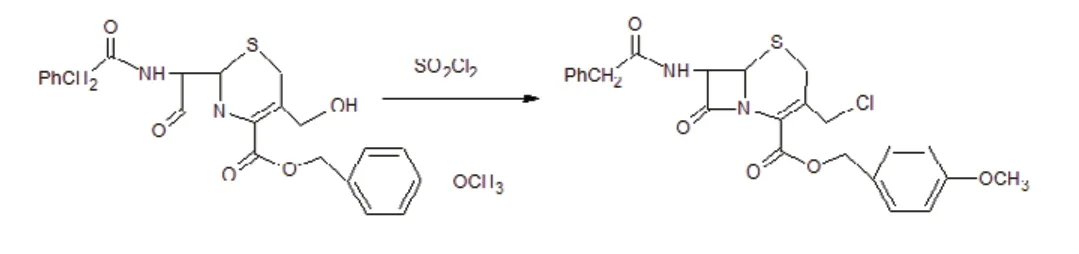
3、3 位 的-CH2OH 卤 化。 将 吡 啶、DMF、SOCl2的混合液加到7- 苯乙酰胺-3- 羟甲基头孢烷烯酸对甲氧基苄酯中,再加入THF 反应10 分钟,减压除溶剂后加乙酸乙酯,然后依次用NaHCO3溶液、水洗涤。有机相用活性炭脱色过滤,滤液用无水硫酸镁干燥。去除溶剂加入二氯甲烷,再用石油醚重结晶,即得产物。反应路线如下:
第二类:扩环前卤代
闭环前进行的卤代主要是通过Morin 转换,将青霉素亚砜酯开环后用捕捉剂捕捉,然后进行烯丙位的氯化,最后闭环得到产品。根据开环所用原料的不同分为三种。
4、2- 巯基苯并噻唑。将青霉素G 对甲氧基苄酯与2- 巯基苯并噻唑在甲苯中回流5h, 去溶剂即得开环产物,然后将其与苯亚磺酸钠溶于丙酮中室温反应4 小时过滤,去滤液用薄板层析得取代产物。反应路线如下:
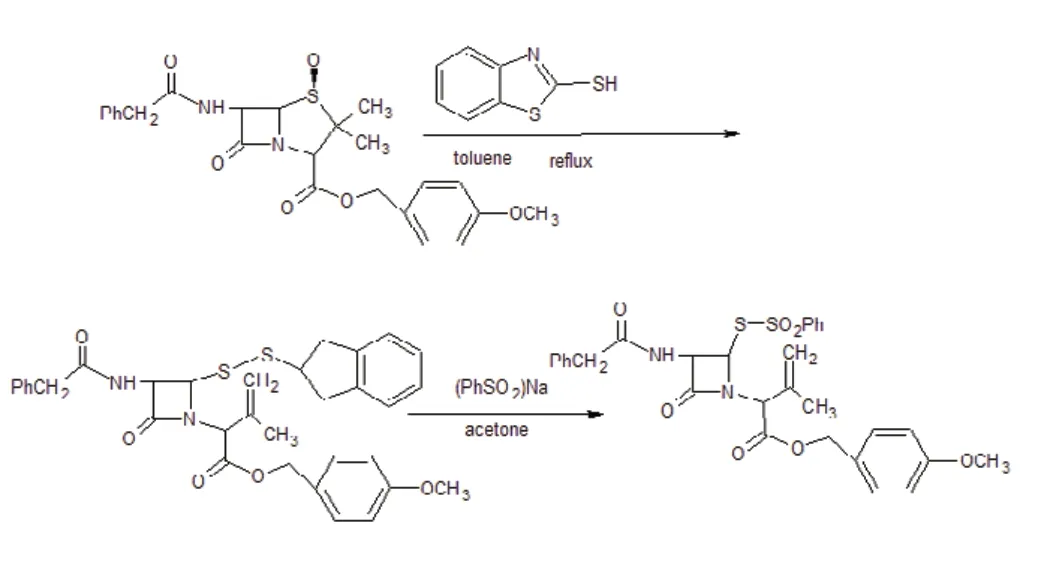
5、 亚磷酸三甲酯
将青霉素G 对甲氧基苄酯与亚磷酸三甲酯反应,生成开环产物。Torris 等将开环产物溶于甲醇,加入盐酸,再滴加苯磺酰溴,反应混合物分别用盐水和乙酸乙酯洗涤。有机相用无水硫酸镁干燥,用二氧化硅薄层分离,得到最终产品。反应路线如下:

6、 苯亚磺酸盐
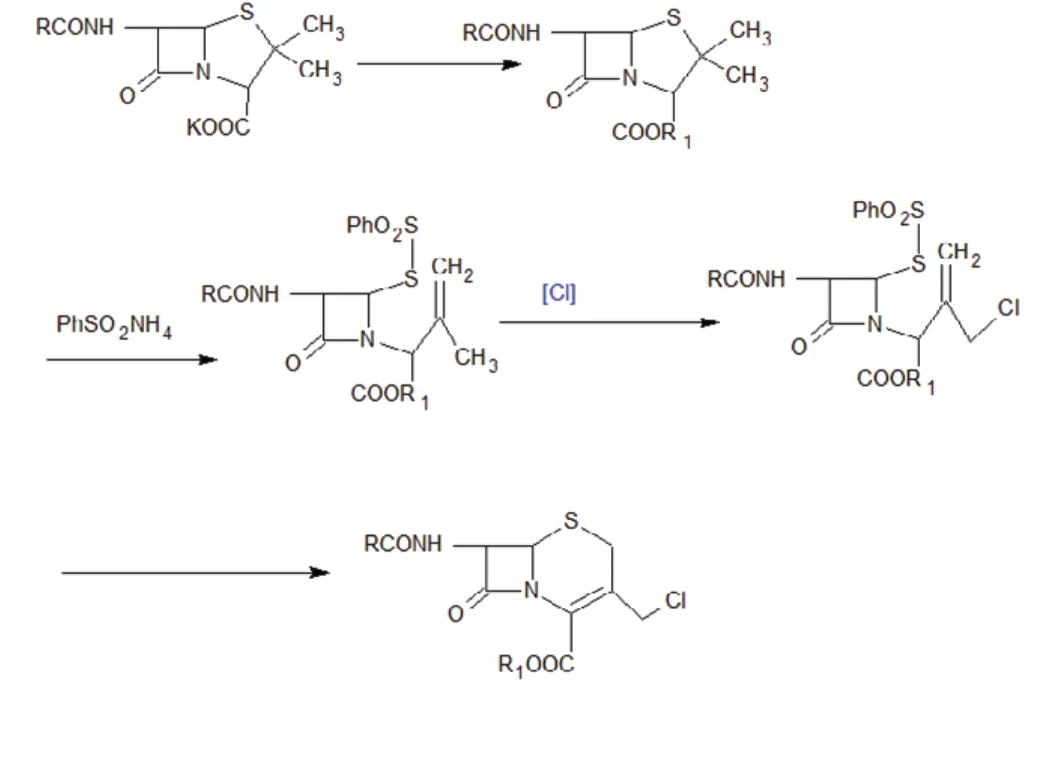
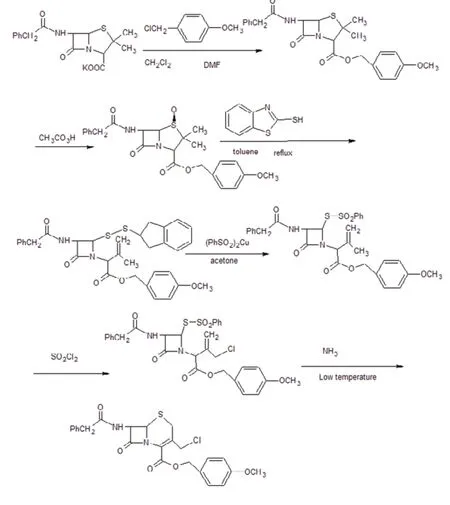
即以青霉素为原料,经过氧化、酯化形成青霉素亚砜酯后,再与苯亚磺酸盐进行反应,开环生成氮杂丁酮亚磺酸中间体,然后用电解含有硫酸的饱和食盐水得到的氯化剂进行氯化,最后氨解闭环,生成GCLE。反应路线如下:
三、结语。
这些合成路线中,路线1 反应步步骤过多,收率太低;路线2 成本高;路线3 起始原料不易获得;路线5 操作较复杂,反应中需使用毒性较大的亚磷酸三甲酯,且储存需要用氮气保护,以防自动氧化生成磷酸三甲酯;且路线4 和5 都用层析法拿出产品,不易形成规模化生产;路线6 使用电解含有硫酸的饱和食盐水得到的氯化剂进行氯化,条件不易控制。我们结合路线4 和6,采用2- 巯基苯并噻唑为开环试剂,利用路线6,采用氯化亚砜为氯化剂,这样就会使GCLE 的合成路线短、原料易得、收率较高和操作相对简单等优点。反应路线如下:
[1]徐俊英,肖鉴谋,章茹等,青霉素合成新型头孢菌素中间体-GCLE 的研究[J].江西化工,2004,7(2):60-63。
[2]赵美法,头孢中间体GCLE 的生产现状与发展[J]。化工科技市场,2005,11(7):37-41。
[3]Kukolja S,Lammert S R,Gleissner M R B,et al.A rear-rangement of penicillin sulfoxides to 3-methylenecephams via sulfinyl intermediates[J].Amer Chem Soc,1976,98(16):5040
[4]Ta-Sen Chou.Process for preparing a 3-exomethylenecepham sulfoxide from penicillin sulfoxides:US 4075203[P].1978-02-21.
[5]T a-Sen Chou.Process for preparing a 3-exo methy lenece phamsul foxides:US 4190724[P].1980-02-26.
[6]Ta-Sen Chou.Process for preparing 2-chlorosulfinylaze-tidinones:US 4289695[P].1981-09-15.
[7]Koppel G A.Kinnick M D,Nummy L J.The conversion of 3-exo-methylene cephalosporin to 3-halomethylcephems;a converient synthesis of 3-substituted cephalosporins from penicillins[J].Amer Chem Soc,1977,99(8):2822-2823.
[8]Dunn M W,Berkowitz F E,Miller J J,et al.Hepatosplenic cat-scratch disease and abdominal pain[J].Pediat Infect Dis,1997,16(3):1269.
[9]Rolf Angerbauer,Michael Boberg,Karl G,et al.Novel β-lactam antibiotics:US,4632918[P].1986-12-30.
[10]Torri S,Hideo Tanaka,Takashi S,et al. Electrochemical S-S bond Fisson of 4-(2-benzothiazolyldithio) azetidinoness[J].Chemistry letters,1982,131(11):1829-1832.
[11]Torri S,Hideo Tanaka,Nobuhito Tada,et al.Ene-type chlorination of olefins with dichlorine monoxide[J].Chemistry Letters,1984,150(6):877-880.
[12]Torri S,Hideo Tanaka,Masa to shi Taniguchi,et al.Deprotection of carboxylic esters of β-lactam homologues;cleavage of p-methoxy benzyl,diphenylmethyl and tertbutyl esters effective by a phenolic matrix[J].J Org Chem,1991,156(11):3633-3637.
[13]Takashi Kamiya,Tsutomu Teraji,Yoshihisa Saito,et al.Studies on β-lactam antibiotics I;A novel conversion of penicillins into cephalosporins[J].Tetrahedron Letters,1973,14(32):3001-3004.
[14]Udayampalayam,Palanisamy.An improved process for the preparation of chloromethyl cephem derivatives:WO,200439813[P].2004-05-13.
[15]Yoshioka M.Synthetic studies related to oral β-lactam antibiotics[J].Pure Apple Chem,1987,59(8):1041-1046.
猜你喜欢
燃料化学学报(2022年5期)2022-05-30
中国药学药品知识仓库(2022年10期)2022-05-29
汕头大学学报(自然科学版)(2020年4期)2020-12-14
猪业科学(2018年5期)2018-07-17
国外医药(抗生素分册)(2016年1期)2016-07-10
合成化学(2015年4期)2016-01-17
海军航空大学学报(2015年1期)2015-11-11
股市动态分析(2015年12期)2015-09-10
中国当代医药(2015年22期)2015-03-01
中国当代医药(2015年7期)2015-03-01
