
散发性肌萎缩侧索硬化症起病部位和进展模式及其相关影响因素分析
2015-02-18 12:23:58党静霞靳娇婷胡芳芳
西安交通大学学报(医学版) 2015年4期
党静霞,靳娇婷,胡芳芳,贾 蕊
(西安交通大学医学部第一附属医院神经内科,陕西西安 710061)
肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)是一种选择性侵犯神经系统上运动神经元(upper motor neuron,UMN)和下运动神经元(lower motor neuron,LMN)的致命性神经系统变性病。临床表现为逐渐加重的延髓或肢体肌肉无力、萎缩,最终影响呼吸肌导致呼吸衰竭而死亡。本病绝大多数为散发性。由于其发病模式和疾病不同阶段的症状组合不同,导致其临床表现多样性,并且起病后疾病进展速度及进展模式也有所不同。通常本病平均存活时间为3~5年,但有10%的患者生存时间超过5年,有5%的患者甚至达到10年[1]。尽管患者的临床表现和病情进展存在较大的个体差异,但本病无一例外均呈现进行性加重。由于本病目前尚无有效治愈方法,因此,当确诊后了解首发起病部位与起病后疾病进展顺序和进展速度之间的关系及影响因素,对于临床新药研发、评估患者预后及使患者及家属对确诊后的生活有一个合理的安排,将提供指导。因此,我们前瞻性设计了此项研究,对158例散发性ALS患者,通过患者自己描述,详细记录患者的首发起病部位,又通过对患者及家属的随访,详细了解患者起病后疾病的进展情况,以期寻找首发起病部位与起病后疾病进展率及疾病进展模式之间的关系及其影响因素。
1 资料与方法
1.1 患者纳入条件 共纳入了2009年1月~2014年8月在西安交通大学医学部第一附属医院神经内科门诊或住院部确诊,并且资料完整的散发性ALS患者158人。所有患者的诊断是由我科ALS专科医生确定,均符合修正后EI Escorial诊断标准[2]。上、下运动神经元损害体征判断按照以下要求:肢体下运动神经元损害的体征包括肌肉萎缩、无力、肌束震颤及腱反射消失。肢体上运动神经元损害的体征包括肢体肌张力增高、腱反射活跃或亢进、髌或踝阵挛阳性、病理征阳性。延髓损害的下运动神经元体征包括舌肌萎缩、纤颤、软腭活动及咽反射消失;延髓损害的上运动神经元体征包括下颌反射亢进及病理征阳性。
1.2 患者信息采集 对所有确诊患者除收集患者的一般资料外,还包括首发起病部位及时间,次发起病部位及时间,病程进展到其他体区的顺序及时间。所有患者均能够较为准确说出首发起病部位。首发起病部位和起病后疾病进展顺序是按照患者及家人的描述,将临床上首先被累及的体区按照延髓、颈髓和腰髓出现相应的症状和体征来确定,如果确诊时仅有一个体区受累,则需要对患者每半年进行1次随访,直至出现第二个体区受累的症状,以确定患者起病后疾病进展的顺序。随访的形式主要为电话随访,部分患者为亲自来访。起病部位处上、下运动神经元损害体征根据对患者进行神经系统查体确定。
对所有纳入患者都按照国际统一的ALSFRS-r量表进行疾病功能状态评分,总分为48分,分值越高,神经功能损害越轻,分值越低,神经功能损害越重。疾病进展率=(基线评分-随访时评分)/随访间期(月)。当疾病进展率>1.0,为快速进展型,当疾病进展率≤1.0,为进展较慢型。本研究调查内容经过医院伦理委员会审核通过,所有接受此项研究的患者均签署了知情同意书。
1.3 首发起病部位分型 首发症状表现为言语不清,饮水呛咳,舌僵硬及舌肌萎缩者为延髓型;首发症状表现为上肢无力及萎缩者为颈髓型;首发症状表现为下肢无力及萎缩者为腰髓型。
1.4 进展类型 按照患者临床上从首发起病部位扩展到身体其他体区进展的顺序,具体分型内容见表1。

表1 进展类型定义Tab.1 Definition of spreading patterns
1.5 统计学分析 采用SPSS V20分析检测数据。各组间计量资料以均值±标准差(±s)表示,对各组间关系采用交叉表及卡方检验进行分析,当交叉表数据符合标准检验条件时,采用Person卡方检验值,当交叉表数据不符合标准检验条件时,采用Fisher精确概率法检验值,P<0.05为组间差异有统计学意义。
2 结 果
2.1 一般结果 共158人,其中男92例(58.2%),女66例(41.8%),男女之比1.39∶1。从发病到确诊的时间为(15.1±12.7)(3~74)月。平均起病年龄为(52.87±10.21)(30~79)岁。年龄小于40岁19例(12%),介于40~60岁之间105例(66.5%),大于60岁34例(21.5%)。首发起病部位局限在延髓及单侧上或下肢者共149例,占94.3%,其中,延髓起病者26例(16.5%);颈髓起病者94例(59.5%),腰髓起病者38例(24.1%)。
2.2 起病部位与性别、侧别及年龄之间的关系 对起病部位与性别、侧别、年龄分组进行交叉表分析及卡方检验,发现起病部位与性别、侧别之间无关(P分别为0.288和0.498),起病部位和年龄分组有关(Fisher精确检验,P=0.027,表2)。其中,在各年龄组中,颈髓起病者都是最多;而在40岁以下患者中,延髓起病较少;在60岁以上患者中,腰髓起病者较少。

表2 起病部位和年龄分组间的关系Tab.2 Relationship between disease-onset regions and age groups [n(%)]
2.3 起病部位处上、下运动神经元损害体征情况对所有患者进行数据分析,发现仅有1例患者首发起病肢体表现为上运动神经元损害体征,其余157例(99.36%)首发起病部位都具有下运动神经元损害体征表现,其中仅有单纯下运动神经元损害体征者68例(43.3%),合并上运动神经元损害体征者为89例(56.3%)。对首发起病部位处上、下运动神经元损害体征与起病部位进行交叉表分析及卡方检验,发现首发起病部位处上、下运动神经元损害体征与起病部位有关(P=0.000);延髓起病者多表现为下运动神经元损害体征(65.4%);颈髓起病者多以一侧上肢单纯下运动神经元损害(47.9%)或合并上运动神经元损害(52.1%)体征为主;而腰髓起病者主要以一侧下肢上、下运动神经元混合损害体征为主(83.8%,表3)。
2.4 进展类型 在158例患者中,有7例患者在截至随访结束时尚未出现第二个部位损害的症状,因此,仅对151例患者的进展类型进行分析。其中延髓起病者26例,其进展类型分别为:垂直型进展到颈髓即嘴端-尾端21例(80.8%),孤立型3例(11.5%),非连续进展到腰髓2例(7.6%)。肢体起病者125例,对进展类型和首发起病部位进行交叉表分析和卡方检验,发现进展类型和首发起病部位有关(Fisher精确检验,P=0.040,表4)。颈髓起病者以循环型、水平型和垂直型进展较多见;而腰髓起病者以循环型进展较多见。
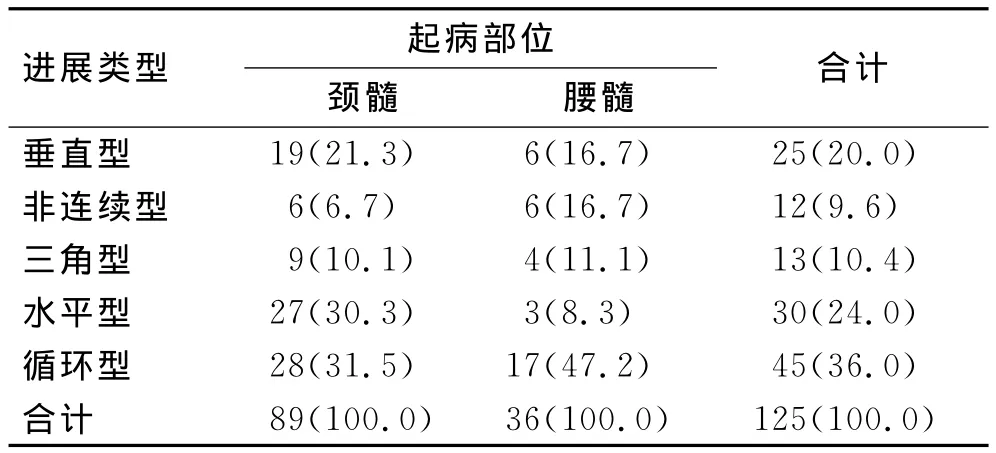
表4 肢体起病和进展类型之间的关系Tab.4 Relationship between limb onset and spreading patterns [n(%)]
2.5 进展率与起病部位、年龄及进展类型之间关系对进展率和起病部位、进展类型及起病年龄分组进行交叉表分析和卡方检验,发现进展率和起病部位及进展类型之间不相关(Fisher精确概率检验,P值分别为0.593和0.091),进展率和起病年龄分组相关(P=0.011,表5)。其中60岁以下起病者进展较慢,60岁以上起病者进展较快。

表5 进展率和起病年龄之间的关系Tab.5 Relationship between rate of progression and age groups [n(%)]
3 讨 论
有关ALS运动系统真正起始发生变性的部位至今仍不清楚。CHARCOT首次描述ALS患者的脊髓前角细胞即下运动神经元损害是继发于脊髓侧索变性所致,并由此认为ALS首发起病部位应该在上运动神经元即大脑皮层运动区。EISEN等[3]也提出ALS是原发于大脑皮层运动区损害,并进而顺行性进展而损害脊髓前角细胞。CHOU和NORRIS[4]又提出了逆行性损害的假说,认为首先损害脊髓前角,然后逆行向上损害大脑皮层。近来又有学者提出ALS首发部位可能在外周,包括肌肉和神经肌肉接头[5]。虽然学者们对ALS首发起病部位争议很大,但研究公认其首发病变部位是局限在运动系统的某个部位,并且起病部位是随机的[6-7]。本研究也证实了这一点,158例患者中首发起病部位局限在延髓或单侧上或下肢者共149例,占94.3%。其中,颈髓起病者最多,为94例,腰髓起病者38例,延髓起病者最少,为26例。本研究结果显示其首发起病部位与性别无关,表明性别并不影响ALS的首发起病部位。首发起病部位与侧别之间也无明显关系,表明首发起病部位对侧别没有明显的倾向性。同时,本研究通过对首发起病部位和起病年龄分组之间的关系进行分析,发现在各年龄组中,颈髓起病者都是最多;而在40岁以下患者中,延髓起病较少;在60岁以上患者中,腰髓起病者较少。虽然我们尚无法解释上述现象,但首发部位的局限性提示ALS早期运动神经元的变性是从脊髓或大脑皮层运动轴的某个局部开始的,肢体起病多于延髓起病及首发起病部位在起病年龄上的差异可能是多因素相互作用导致某些部位在某个年龄段的易感性和抵抗性所致。
国外研究认为,虽然绝大多数ALS患者首发起病部位是局限的,但由于上、下运动神经元在其各自神经解剖学通路上变性损害的程度可能不一样,因此,首发部位在不同体区损害,表现出来的上、下运动神经元损伤的体征差异却很大[8]。有以下运动神经元损害为主的ALS,有以上运动神经元损害为主的,还有以延髓损害为主的,有极少一部分ALS患者是以呼吸肌麻痹为首发症状的。本研究通过对起病部位处上、下运动神经元损害分布情况分析,发现在首发体区起病时,几乎所有患者均以下运动神经元损害症状起病,仅有1例患者发病时表现为一侧下肢僵硬,直到2月后才出现肌肉无力和萎缩。而且首发部位处上、下运动神经元损害的体征也不一样。其中,延髓起病者以下运动神经元损害体征居多;腰髓起病者单纯出现下运动神经元损害体征很少,主要表现为上、下运动神经元损害体征同时存在。国外的研究[9-10]也发现了这种现象。这提示早期支配延髓区的下运动神经元及支配下肢的大脑皮层运动区的上运动神经元可能极容易发生变性,导致以延髓损害起病者,早期上运动神经元损害的体征很难发现,而以一侧下肢即腰髓起病者,早期上运动神经元损害的体征可能较为突出。由于ALS患者首发症状是由患者第一个感知的,并且早期病灶局限,存在的相互干扰因素较少,因此,患者的主诉及对首发部位体区上、下运动神经元损害的体征检查对诊断早期ALS具有重要的临床意义。
影响ALS病程进展的因素可能很多,包括起病部位、年龄及进展类型等。多数研究认为其进展具有一定的方向性,而非无规律性。EISEN等[11-12]对一组患者的运动缺失症状进行了横向研究,发现在疾病早期,上、下运动神经元损害是相互关联的,起于同一体区,但之后的进展方式不同,上和下运动神经元的进展是沿着各自的解剖学通路进展的,但他们不能解释其他的传播类型,其进展模式的准确发病机制也不清楚。RAVITS[13-14]认为起始运动神经元的变性是在脊髓轴的某个准确部位,然后沿着神经轴的三维方向向上、下及向对侧进展,其扩展是有方向性的,它和运动神经元池的走向、首发病灶与邻近节段的距离、局部节段运动神经元的易感性等有关。SONJA等[10]研究发现ALS进展首先是沿着相邻的体区发展,然后再影响身体其他部位。本研究结果显示,ALS患者延髓起病者以嘴端-尾端垂直型即向下发展到颈髓者较多,而跨区域非连续进展到腰髓的很少;颈髓起病者以循环型、水平型和垂直型进展较多;而腰髓起病者以循环型进展较多;各型进展中以非连续型进展最少见。可以看出,ALS起病后疾病进展的方向是不一样的,有水平向进展,也有从尾端到嘴端或嘴端到尾端进展,还有沿着脊髓轴循环进展,不同的进展方向导致其临床表型的多样性。但不管进展方向如何,其进展多数是沿着相邻体区发展的,而跨体区的非连续型进展很少见,这也从另一个侧面反映出ALS发病的不同分子机制及病理过程的复杂性。本研究同时也发现进展率和起病部位及进展类型之间无明显相关。但进展率和起病年龄之间有关,60岁以下起病者的进展相对较慢,60岁以上起病者的进展较快。即发病年龄可能是ALS预后的一个重要影响因素,但究竟是年龄本身还是年龄外的其他因素导致,还需要进行更深入的研究。
[1]TURNER MR,PARTON MJ,SHAW CE,et al.Prolonged survival in motor neuron disease:a descriptive study of the Kings’database 1990—2002[J].J Neurol Neurosurg Psychiatry,2003,74(7):995-997.
[2]BROOKS BR,MILLER RG,SWASH M,et al.El Escorial revisited:revised criteria for the diagnosis of amyotrophic lateral sclerosis[J].Amyotroph Lateral Scler Other Motor Neuron Disord,2000,1(5):293-299.
[3]EISEN A,KIM S,PANT B.Amyotrophic lateral sclerosis(ALS):aphylogenetic disease of the corticomotorneuron?[J].Muscle Nerve,1992,15(2):219-224.
[4]CHOU SM,NORRIS FH.Amyotrophic lateral sclerosis:lower motor neuron disease spreading to upper motor neurons[J].Muscle Nerve,1993,16(8):864-869.
[5]FISHER LR,CULVER DG,TENNANT P,et al.Amyotrophic lateral sclerosis is a distal axonopathy:evidence in mice and man[J].Exp Neurol,2004,185(2):232-240.
[6]RAVITS J,PAUL P,JORG C.Focality of upper and lower moor neuron degeneration at the clinical onset of ALS[J].Neurology,2007,68(19):1571-1575.
[7]GORDON PH.Amyotrophic Lateral Sclerosis:An update for 2013 Clinical Features,Pathophysiology, Management and Therapeutic Trials[J].Aging Dis,2013,4(5):295-310.
[8]GARGIULO-MONACHELLI GM,JANOTA F,BETTINI M,et al.Regional spread pattern predicts survival in patients with sporadic amyotrophic lateral sclerosis[J].Eur J Neurol,2012,19(6):834-841.
[9]ZOCCOLELLA S,BEGHI E,PALAGANO G,et al.Signs and symptoms at diagnosis of amyotrophic lateral sclerosis:apopulation-based study in southern Italy[J].Eur J Neurol,2006,13(7):789-792.
[10]SONJA K,KATJA K,MARION F,et al.Onset and spreading patterns of upper and lower motor neuron symptoms in amyotrophic lateral sclerosis[J].Muscle Nerve,2011,43(10):636-642.
[11]EISEN A,WEBER M.The motor cortex and amyotrophic lateral sclerosis[J].Muscle Nerve,2001,24(4):564-573.
[12]EISEN A.Amyotrophic lateral sclerosis-evolutionary and other perspectives[J].Muscle Nerve,2009,40(2):297-304.
[13]RAVITS J,LAURIE P,FAN Y,et al.Implications of ALS focality:rostral-caudal distribution of lower motor neuron loss postmortem[J].Neurology,2007,68(19):1576-1582.
[14]RAVITS J,ABERT R,SPADA LA.ALS motor phenotype heterogeneity,focality and spread[J].Neurology,2009,73(10):805-811.
猜你喜欢
中国实用神经疾病杂志(2020年19期)2020-10-23 06:53:44
考试与评价·高二版(2020年2期)2020-09-10 07:22:44
中风与神经疾病杂志(2020年7期)2020-08-06 13:25:02
医学与法学(2020年2期)2020-07-24 08:46:48
河北医科大学学报(2017年2期)2017-02-23 18:58:29
中外医疗(2015年16期)2016-01-04 06:51:42
微生物与感染(2015年5期)2015-12-08 07:03:24
实用手外科杂志(2015年1期)2015-08-27 01:52:06
中国康复理论与实践(2015年7期)2015-05-09 08:31:51
哈尔滨医药(2014年2期)2014-02-27 13:35:03
