
慢性肾脏病动脉中层钙化细胞生物学机制研究进展
2015-02-07 13:51:54孙旦芹宋丹杨俊伟何伟春
中国循证心血管医学杂志 2015年4期
孙旦芹,宋丹,杨俊伟,何伟春
• 综述 •
慢性肾脏病动脉中层钙化细胞生物学机制研究进展
孙旦芹,宋丹,杨俊伟,何伟春
慢性肾脏病(CKD)是公认的“全球公共健康问题”,我国成人CKD的患病率已达10.8%[1]。随着肾功能进行性恶化,心血管疾病(CVD)成为CKD患者的主要死亡原因之一,而血管钙化是其中最常见的病理表现,血管钙化导致的死亡率约占终末期肾脏病(ESRD)总病死率的30%左右[2]。动脉中层钙化是慢性肾脏病患者血管钙化的主要类型,血管平滑肌细胞(VSMCs)是动脉中层的主要成分,慢性肾脏病的多种危险因素如高磷、高钙、炎症等可诱导VSMCs发生自噬、凋亡及表型改变,对血管钙化的发生起了重要作用[3]。本文主要就慢性肾脏病环境中VSMCs细胞生物学变化在血管钙化发生发展中作用的研究进展作一综述。
1 慢性肾脏病血管钙化的病理学特点
血管钙化主要有两种病理类型,一种是内膜钙化,即动脉粥样硬化,基本病变是动脉内膜的脂质沉积,粥样斑块形成,致管壁变硬、管腔狭窄等;另一种是中层钙化,以羟基磷灰石晶体钙沿着动脉中层弥漫沉积于整个血管壁为特征,致动脉中层厚度增加,管壁僵硬,顺应性降低,这种动脉中层钙化是慢性肾脏病患者最常见的血管钙化类型[4]。在钙化的动脉中层有许多骨相关蛋白的表达,包括骨钙素(OC)、骨桥蛋白(OP)和骨保护素(OPG),以及多种骨形态发生蛋白(BMPs),它们均参与了动脉的钙化重塑过程[5]。
2 血管平滑肌细胞的病理改变是慢性肾脏病血管钙化发生的关键因素
近年来随着血管钙化机制研究的不断深入,目前已知许多因素能够促进CKD血管钙化,比如遗传、高龄、糖尿病、高血压、吸烟等传统的因素,非传统的危险因素包括透析时间、氧化应激、高磷、高钙、炎症状态及毒素环境等。在这些影响因素中,CKD患者的血管钙化与钙磷调节紊乱密切相关,高磷、钙磷乘积升高可直接影响血管平滑肌细胞的功能,从而参与CKD患者心血管并发症的发生发展。
现在公认观点是,慢性肾脏病患者的血管钙化并不是简单的钙磷沉积的被动过程,而是VSMCs在CKD的环境中经历凋亡和囊泡形成并向成骨样细胞表型改变,进而诱导基质形成、合成钙化蛋白并吸引钙磷沉积的主动过程[6,7]。超微结构分析也证实,高磷环境中VSMCs表面出现含有磷灰石的基质囊泡和钙化的胶原纤维,基质囊泡为钙化提供晶核形成的位点,与成骨细胞表面芽生的囊泡极为相似[8]。VSMCs的病理改变在慢性肾脏病患者血管钙化中起关键作用。
3 CKD环境下血管平滑肌细胞的表型改变
3.1 VSMCs表型转化的特征VSMCs是一种非终末分化细胞,其表型具有可塑性。在高钙、高磷、高糖、炎症、氧化应激等的作用下,VSMCs会发生表型转化,由主要执行舒缩功能的收缩表型转变为合成表型,获得合成钙化相关蛋白的能力,对血管钙化起关键性作用[5,7]。体外实验表明,高磷环境能够使VSMCs丢失平滑肌细胞标志分子如平滑肌22α(SM22α)、α-平滑肌肌动蛋白(α-SMA)等,干扰VSMCs中的微丝重构并抑制细胞的运动和收缩功能;同时,高磷促使VSMCs表达成骨细胞标志分子Runx2/Cbfα1、Msx2、OSX(osterix)等[6,8],使细胞向成骨样细胞表型转化。Runx2/Cbfα1是一种成骨细胞特异性核转录因子,对成骨细胞分化、骨基质基因表达以及骨矿化都是必须的,Yabing et等[9]报道特异性血管平滑肌细胞Runx2基因敲除的小鼠能部分抑制血管钙化;Msx2基因是同源基因Msh家族成员,在骨形成和成骨细胞分化过程中发挥重要作用,Msx2的大量表达可以引起下游的另一个骨转录因子OSX表达增加;OSX是由Nakashima等[10]发现的成骨细胞分化终末期的一种重要的特异性转录因子,只在发育的骨组织中特异性表达,是骨矿化过程中所必需的转录因子,OSX自身的突变或缺失也可导致骨矿化的迟滞或停止。这些成骨细胞核特异性转录因子在VSMCs中的表达增加后可促进许多骨基质成分的表达,如I型胶原、OC、OP以及碱性磷酸酶(ALP)等的表达。I型胶原能够促进钙化结节的形成加速,增加钙的侵入,OC和OP均是骨基质中调节矿化的非胶原蛋白,而ALP是早期调节骨矿化的关键酶,其升高提示存在高的骨形成率[5]。这些基质蛋白表达增加后可作为其后矿化的核心,使钙盐和骨基质蛋白沉积于动脉中层(图1)。
3.2 高磷诱导VSMCs表型转化的机制临床研究证实,循环中高磷是血管钙化的一个独立危险因素,当血清磷高于6.5 mg/dl时能够显著地增加心血管事件的风险,并增加ESRD患者的死亡率[11]。动物实验表明[12],7/8肾切除CKD小鼠饲以高磷饮食8周后,主动脉中层有钙盐沉积。
体外研究发现[13],无机磷(P的浓度>1.4mmol/L)对VSMCs周围基质矿化具有剂量依赖性。此种效应依赖于细胞外高磷能够激活VSMCs细胞膜上的III型钠-磷协同转运通道(NPC)中的Pit-1,增加磷在细胞内的水平,继而诱导Runx2/Cbfα1等核转录因子的表达,启动血管钙化过程的发生。当将NPC抑制剂磷甲酸(PFA)与VSMCs共培养时,VSMCs对细胞外磷的摄取受到抑制,进入细胞内的磷减少,继而影响Runx2/Cbfα1表达的启动,从而预防钙化的发生。近期研究表明[14],高磷环境下的VSMCs以及高磷饮食的尿毒症大鼠主动脉组织中,Klf4因子的mRNA及蛋白的表达明显增高,进一步研究发现VSMCs中的Klf4因子在高磷诱导下能够结合到骨转录因子的启动子区域,调节VSMCs成骨基因的转录,促进成骨细胞相关蛋白表达增加。因此,在高磷环境下,经过膜通道转运进入VSMCs内的磷通过启动成骨细胞转录因子表达促使VSMCs向成骨样表型转化。
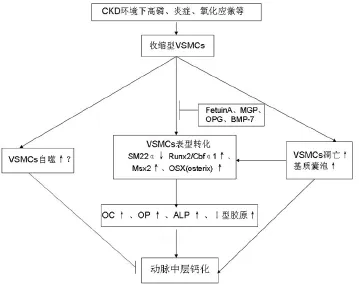
图1 动脉中层钙化的发生过程
关于高磷如何启动和调控VSMCs成骨调节和生物矿化程序的关键细胞内信号途径,目前仅有部分报道。我们研究发现高磷能够活化VSMCs内转化生长因子(TGF)-β,并上调其I型受体(TβRI)的表达,TGF-β1的中和抗体能明显拮抗高磷对VSMCs的生物学效应,提示TGF-β1直接介导了细胞的表型转化过程[15]。Julio M等证实在培养的VSMCs中,高磷通过活化Wnt/β-catenin信号通路增加Runx2/ Cbfα1表达,促进VSMCs向成骨样细胞表型转化;而活化帕立骨化醇通过抑制VSMCs内Wnt/β-catenin信号通路的活化,抑制高磷诱导的Runx2表达从而阻止细胞表型转化[16]。体内研究表明[12],在高磷饮食诱导尿毒症大鼠血管钙化的晚期,分析主动脉组织中的基因发现,Wnt拮抗剂——分泌性Frizzled相关蛋白1、2、4(sFRP1、sFRP2和sFRP4)均上调,表面上看这似乎与以往认为的Wnt信号促进钙化的观点相矛盾,然而它也提示,升高的sFRPs很可能是防御机制的一种表现,目的是减弱或阻断过度活跃的Wnt信号,进而减少动脉壁的矿化以避免血管钙化的进一步发展。近期一项研究[17]发现高磷可以通过增加线粒体膜电位而促进线粒体活性氧(ROS)的产生增加,进而活化致炎转录因子NF-κB信号通路,促进收缩表型的VSMCs向成骨样细胞表型转化,促进血管钙化的发生。此外,ERK信号通路、P38/MAPK信号通路也参与磷所诱导VSMCs表型转化的过程,具体机制有待进一步阐明[18]。
3.3 VSMCs表型转化的抑制因素在CKD环境下促钙化动力因素与抑制钙化因素不平衡的综合结果也是血管钙化发生的原因之一。促进钙化因素有:年龄、钙磷代谢紊乱、糖尿病、炎症以及透析年龄等。抑制因素有:胎球蛋白(Fetuin A)、基质GLA蛋白(MGP)、骨保护素(OPG)、骨形成蛋白-7(BMP-7)、焦磷酸盐、骨桥蛋白等。其中Fetuin A、MGP以及OPG等不仅能抑制钙磷沉积还能抑制VSMCs表型转化[5]。Fetuin A是一种糖化蛋白,主要由肝脏合成和分泌进入循环,聚集于骨骼中,为细胞外钙调节蛋白,能与钙磷形成高分子量复合物,抑制过饱和的钙磷盐沉积;此外,Fetuin A分子的氨基末端存在与TGF-β II受体相似的序列,可与TGF-β结合抑制VSMCs向成骨样细胞转分化,避免钙化进展。Ketteler等[19]报道尿毒症患者血清Fetuin A普遍降低的,且低Fetuin A与透析患者心血管死亡率和全因死亡率增加相关。MGP在血管中主要由VSMCs 产生的细胞外基质蛋白,含有5个维生素K依赖性的谷氨酸残基,是一种维生素K依赖性蛋白。维生素K能促使MGP的谷氨酸残基转变成羧基化谷氨酸残基,羧基化能使MGP蛋白发挥生理活性,抑制VSMCs的表型转化,并且抑制VSMCs与基质囊泡的钙盐相结合,从而抑制血管钙化进展。华法林是双香豆素类,能够抑制维生素K环氧化物还原酶的活性从而阻断还原型维生素K的生成,影响MGP羧基化反应;尿毒症大鼠应用治疗剂量华法林后能够显著减少体内MGP水平并加重大动脉钙化[20]。OPG是肿瘤坏死因子受体超家族成员,不但能抑制VSMCs表型转化,还能够抑制动脉组织ALP活性,抑制钙化进展[2,4,5]。随着慢性肾脏病的进展,营养状况、钙磷紊乱、炎症状态等情况加重,这些钙化抑制蛋白消耗增多和表达不断下降。因此,促进VSMCs表型转化因素的增强与细胞主动防御机制的削弱导致慢性肾脏病患者的血管钙化进行性加重。
4 血管平滑肌细胞的凋亡
基质囊泡是由增殖的软骨细胞或骨细胞浆膜形成的独立于细胞外的细胞器。基质囊泡的膜富含类脂并具有很高的ALP活性,可水解基质中多种磷酸酯,使无机磷浓度升高;囊泡中富含丝氨酸磷酯,具有富集钙离子生成无定形磷酸钙并进一步转化为磷灰石的作用。胚胎骨、软骨及牙本质中的矿物都是通过基质囊泡在细胞外形成的。
近来研究发现,VSMCs发生凋亡或坏死后的降解产物—基质囊泡,是血管钙化的始动环节或起始点[21,22]。VSMCs来源的基质囊泡是矿化竞争的一种方式,目的是减少细胞内过多的钙负荷带来的细胞毒性损伤,在正常机体血管壁中,这些囊泡会被吞噬细胞及时吞噬并降解,不会引起动脉中层矿化。但在CKD状态下,各种致钙化因素的刺激导致VSMCs受损后,含有很高ALP活性的囊泡从活的或者即将死亡的VSMCs中释出,形成碱性钙磷沉积的微环境,增加羟基磷灰石沉积,同时基质囊泡还能结合细胞外的基质蛋白如OC、OP、I型胶原等从而启动血管钙化过程[23,24]。
Shroff RC等[25]报道透析相关因素如血流动力学改变等会增加患者血管壁VSMCs凋亡,凋亡小体形成后使得这些部位血管壁ALP活性明显增高形成易于钙化沉积微环境,囊泡增多后还可以富集大量钙离子,高钙反过来又加重细胞凋亡,并减少基质囊泡中钙抑制物如MGP和Fetuin-a 的水平,如此恶性循环加重血管钙化。体外研究表明[26],在培养的人VSMCs中,高磷能够影响VSMCs线粒体的代谢途径从而促进VSMCs凋亡;而HMG-CoA抑制剂他汀类药物能够缓解高磷所诱导的凋亡从而部分抑制钙化。临床研究显示,透析后患者桡动脉中膜存在VSMCs凋亡小体,且凋亡发生在血管钙化之前,用他汀类药物干预后能够抑制凋亡并缓解钙化程度[5,19]。因此,高磷、高钙、血液透析都能诱导细胞凋亡并加重血管钙化,但具体机制还有待进一步研究。
5 血管平滑肌细胞的自噬
自噬是细胞利用溶酶体降解自身受损的细胞器和大分子物质的过程,是真核细胞特有的生命现象。它是对外源性刺激(营养缺乏、低氧、氧化应激、感染等)的适应性反应,可作为一种防御机制清除受损的细胞器以及代谢产物等,它作为一种细胞死亡程序诱导细胞主动性死亡[27]。
有关VSMCs自噬参与血管钙化的报道还比较少。Somers P等发现早期主动脉瓣退行性变时,平滑肌细胞发生自体吞噬死亡后会释放很多基质囊泡,并趋化炎症因子,启动钙化程序。而近期王宪等报道,用3-MA以及Atg5 siRNA等自噬抑制物能够增加高磷所诱导的人VSMCs的钙化,用VPA等自噬刺激物反而缓解VSMCs钙盐沉积。体内实验腺嘌呤所诱导的CKD大鼠血管钙化的实验数据也证实了自噬能够部分缓解高磷所诱导的大动脉中层钙化。研究者[29]认为VSMCs的自噬能自身部分拮抗磷所诱导的血管钙化。因此,适当的自噬很可能是VSMCs应对高磷等致钙化因素的一种细胞自我保护性机制,而过多自噬则可能导致细胞的死亡。VSMCs自噬与血管钙化的关系仍有很大争议,待进一步研究来阐明。
综上所述,动脉的中层血管钙化是引起慢性肾脏病患者心血管事件高发的重要因素,其中,VSMCs的病理改变是慢性肾脏病血管钙化的核心问题。多种机制,如凋亡、自噬、表型改变等均参与了CKD血管钙化的进展,随着对VSMCs凋亡、自噬、表型转化等调控作用和分子机制研究的不断深入,对CKD患者血管钙化的防治可提供新的思路以及新靶点,对于降低心血管事件死亡风险,改善患者预后具有重要意义。
[1] Zhang L,Wang F,Wang L,et al. Prevalence of chronic kidney disease in china: A cross-sectional survey Lancet[J]. Lancet,2012,379(9818): 815-22.
[2] Cozzolino M,Mazzaferro S,Pugliese F,et al. Vascular calcification and uremia: What do we know[J]. Am J Nephrol,28(2):339-46.
[3] Kendrick J,Chonchol M. The role of phosphorus in the development and progression of vascular calcification[J]. Am J Kidney Dis,2011, 58(5):826-34.
[4] Shroff R,Long DA,Shanahan C. Mechanistic insights into vascular calcification in ckd[J]. J Am Soc Nephrol,2013,24(2):179-89.
[5] Mizobuchi M,Towler D,Slatopolsky E. Vascular calcification: The killer of patients with chronic kidney disease[J]. J Am Soc Nephrol, 2009,20(7):1453-64.
[6] Shroff RC,Shanahan CM. The vascular biology of calcification[J]. Semin Dial, 2007,20(2):103-9.
[7] Moe SM,Chen NX. Mechanisms of vascular calcification in chronic kidney disease[J]. J Am Soc Nephrol, 2008, 19(2):213-6.
[8] Lau WL,Festing MH,Giachelli CM. Phosphate and vascular calcification: Emerging role of the sodium-dependent phosphate cotransporter pit-1[J]. Thromb Haemost,2010,104(3):464-70.
[9] Sun Y,Byon CH,Yuan K,et al. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification[J]. Circ Res,2012, 111(5):543-52.
[10] Nakashima K,Zhou X,Kunkel G,et al. The novel zinc fingercontaining transcription factor osterix is required for osteoblast differentiation and bone formation[J]. Cell,2002,108(1):17-29.
[11] Giachelli CM. The emerging role of phosphate in vascular calcification[J]. Kidney Int,2009,75(9):890-7.
[12] Roman-Garcia P,Carrillo-Lopez N,Fernandez-Martin JL,et al. High phosphorus diet induces vascular calcification, a related decrease in bone mass and changes in the aortic gene expression[J]. Bone,2010, 46(1):121-8.
[13] Jono S,McKee MD,Murry CE,et al. Phosphate regulation of vascular smooth muscle cell calcification[J]. Circ Res,2000,87(7):e10-e7.
[14] Yoshida T,Yamashita M,Hayashi M. Kruppel-like factor 4 contributes to high phosphate-induced phenotypic switching of vascular smooth muscle cells into osteogenic cells[J]. The Journal of biological chemistry,2012,287(31):25706-14.
[15] Wang N,Wang X,Sun B,et al. Role of tgf-beta1 in production of fibronectin in vascular smooth muscle cells cultured under highphosphate conditions[J]. J Nephrol,2013, 26(1):213-8.
[16] Martinez-Moreno JM,Munoz-Castaneda JR,Herencia C,et al. In vascular smooth muscle cells paricalcitol prevents phosphateinduced wnt/beta-catenin activation[J]. Am J Physiol Renal Physiol, 2012,303(8):F1136-44.
[17] Zhao MM,Xu MJ,Cai Y,et al. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphateinduced vascular calcification in vitro and in vivo[J]. Kidney Int, 2011,79(10):1071-9.
[18] Liu Y,Shanahan CM: Signalling pathways and vascular calcification[J]. Front Biosci,2011,16:1302-14.
[19] Ketteler M,Bongartz P,Westenfeld R,et al. Association of low fetuin-a (ahsg) concentrations in serum with cardiovascular mortality in patients on dialysis: A cross-sectional study[J]. Lancet,2003, 361(9360):827-33.
[20] McCabe KM,Booth SL,Fu X,et al. Dietary vitamin k and therapeutic warfarin alter the susceptibility to vascular calcification in experimental chronic kidney disease[J]. Kidney Int,2013, 83(5):835-44.
[21] Kim KM. Apoptosis and calcification[J]. Scanning Microsc,1995, 9(4):1137-75.
[22] Anderson HC. Molecular biology of matrix vesicles[J]. Clinical orthopaedics and related research,1995(314):266-80.
[23] Proudfoot D,Skepper JN,Hegyi L,et al. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies[J]. Circ Res,2000,87(11):1055-62.
[24] Reynolds JL,Joannides AJ,Skepper JN,et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: A potential mechanism for accelerated vascular calcification in esrd[J]. J Am Soc Nephrol,2004,15(11):2857-67.
[25] Shroff RC,McNair R,Figg N,et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis[J]. Circulation, 2008, 118(17):1748-57.
[26] Son BK,Kozaki K,Iijima K,et al. Statins protect human aortic smooth muscle cells from inorganic phosphate-induced calcification by restoring gas6-axl survival pathway[J]. Circ Res,2006, 98(8):1024-31.
[27] Mizushima N,Komatsu M. Autophagy:Renovation of cells and tissues[J]. Cell,2011,147(4):728-41.
R543.5
A
1674-4055(2015)04-0563-03
2015-04-08)
(责任编辑:张灵)
江苏省自然基金项目(BK2012870)
210003 南京,南京医科大学无锡二院(孙旦芹,宋丹);南京医科大学第二附属医院(孙旦芹,杨俊伟,何伟春)
杨俊伟,E-mail:jwyang_nj1980@hotmail.com
10.3969/j.1674-4055.2015.04.47
猜你喜欢
世界科学技术-中医药现代化(2021年10期)2021-03-02 05:51:18
昆明医科大学学报(2021年1期)2021-02-07 01:06:36
空间科学学报(2020年2期)2020-04-01 03:50:30
安徽医科大学学报(2016年12期)2017-01-15 14:21:53
中国民族医药杂志(2016年6期)2016-05-09 08:52:52
中国医科大学学报(2015年10期)2015-03-01 02:09:58
现代企业(2015年6期)2015-02-28 18:51:22
中国中医药现代远程教育(2014年20期)2014-03-01 04:31:39
中国医学科学院学报(2011年5期)2011-12-01 03:52:51
中国人力资源开发(2011年12期)2011-08-21 09:04:38
