
眼咽型肌营养不良一家系临床与分子遗传学研究☆
2015-02-01 09:20:24陈永洪龙跃生蔡莉莉王海龙马彪傅君毅夏勇李新毅解龙昌
中国神经精神疾病杂志 2015年6期
陈永洪龙跃生蔡莉莉王海龙马彪傅君毅夏勇李新毅解龙昌
·论 著·
眼咽型肌营养不良一家系临床与分子遗传学研究☆
陈永洪*龙跃生*蔡莉莉△王海龙△马彪△傅君毅*夏勇*李新毅△解龙昌*
目的探讨一考虑诊断眼咽型肌营养不良(oculopharyngeal muscular dystrophy,0PMD)家系的临床及分子生物学特点。方法 收集该家系成员的临床资料,并经包括先证者在内的16位家族成员同意,收集其血样进行聚合酶链反应(PCR)基因验证分析。结果该家系成员男性患者起病以眼睑下垂为首发症状,而后开始逐渐出现以发音及吞咽困难为表现的咽部肌群和肢体乏力为表现的四肢近端肌群受累,而女性患者则往往以吞咽困难为首发表现。参与基因检测的家族成员中共发现10位存在多聚腺苷酸结合蛋白核l(PABPN1)基因的(GCG)6重复异常拷贝为(GCG)10,从而导致了丙氨酸的扩增。结论基因诊断及产前诊断是确诊及预防眼咽型肌营养不良的关键,眼睑下垂可能为携带(GCG)10突变男性OPMD患者的首发症状。
眼睑下垂 吞咽困难 眼咽型肌营养不良 多聚腺苷酸结合蛋白核l(PABPN1)基因
眼咽型肌营养不良(oculopharyngeal muscular dystrophy,0PMD)是进行性肌营养不良的一种特殊类型,临床上罕见[1],是以进行性眼睑下垂、吞咽困难和近端肢体无力为主要表现的迟发性遗传性骨骼肌疾病,其在全世界各人种间均有相关报道[2],绝大多数呈常染色体显性遗传,极少数呈常染色体隐性遗传。OPMD患者多于50~60岁左右起病,往往以缓慢进展的眼睑下垂、吞咽困难为首发症状就诊,肌肉病理可见核内细丝样包涵体(intranu⁃clear inclusions,INIs)[3],而这被认为是14号染色体上的多聚腺苷酸结合蛋白核l基因(poly(A)binding protein nucIear l gene,PABPN1)突变所引起的,基因检测发现突变发生于该基因的第一个外显子上,在减数分裂和有丝分裂期异常扩增了(GCG)三核苷酸。在正常的(GCG)6上增加了1~7个(GCG)重复序列,因而成为(GCG)7-13[4],也有部分患者基因型表现为(GCA)的插入,表现为(GCG)6(GCA)n(GCG)n(n=1-4)[5]。由于该病存在成年晚发的特点,早期难以对其明确诊断。现根据我们所掌握家系进行分子遗传学研究,以揭示其分类及其临床特征。
1 资料与方法
1.1 资料 先证者Ⅱ5(见图1)为70岁男性,50岁开始逐渐出现双侧眼睑下垂,55岁开始出现双下肢乏力,62岁开始出现吞咽困难症状,体查示:发育正常,营养中等,讲话带鼻音,构音欠清晰,双侧眼裂变窄,右侧为重,闭合无力,眼球向上活动受限,视物稍模糊,软腭弓上抬受限,咽反射减弱,无饮水呛咳,吞咽动作缓慢,伸舌偏右,舌肌未见明显萎缩,颈部甲状腺软骨上方可见肌肉萎缩后深凹,余肢体肌肉未见明显萎缩,双上肢远端肌力5-级,近端肌力4+级,双下肢远端肌力4+级,近端肌力0级,双下肢感觉减退,腱反射减弱,病理征未引出。辅助检查示:血清肌酸激酶(CK)361 U/L(正常值38~190 U/L),乳酸脱氢酶(LDH)、甲功、免疫指标等均正常。既往肌电图提示存在肌源性改变,新斯的明实验阴性。
家族成员中其他已明确发病的患者包括:Ⅰ
2、Ⅱ1、Ⅱ3、Ⅱ5、Ⅱ7、Ⅱ9、Ⅱ13,其他成员尚未出现确切临床症状。其中Ⅰ2为先证者母亲,50岁开始出现吞咽困难症状,然后逐渐开始出现眼睑下垂及行走困难症状,其80余岁因进食困难及反复肺炎离世。Ⅱ150岁开始出现眼睑下垂症状,逐渐发展至吞咽及行走困难,60余岁因车祸离世。Ⅱ350岁开始出现吞咽困难症状,3年后开始出现眼睑下垂,7年后开始出现行走费力症状。Ⅱ753岁开始出现眼睑下垂症状,9年后开始出现吞咽困难及行走费力症状。Ⅱ950岁开始出现眼睑下垂症状,6年后开始出现行走困难症状,尚未出现吞咽困难表现。Ⅱ1350岁开始出现眼睑下垂症状,数年后开始出现行走费力症状,具体不详。
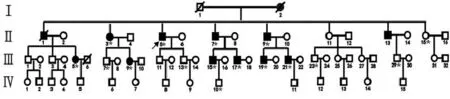
图1 OPMD患者家系系谱图健康男性,健康女性,男性患者,女性患者,先证者,已死亡,*接受基因检测者
1.2 方法
1.2.1 基因扩增 经检查者知情同意,采集包括先证者在内家族共16位成员外周静脉血2 mL于EDTA抗凝管中做基因检查,采用好芝生物公司离心柱法提取血液基因组DNA,通过美国国立生物技术信息中心(NCBI)的GenBank获取PABPN1基因全序列,利用Primer 5.0软件设计引物,上游引物(F):5’-CGCAGTGCCCCGCCTTAGA-3’,下游引物(R):5’-ACAAGATGGCGCCGCCGCCCCGGC-3’,以检查者基因组DNA为模板对目标基因序列进行聚合酶链反应(PCR)扩增,所用试剂均来自TA⁃KARA公司,PCR反应25µL体系包括:基因组DNA(100 ng)1µL、10 mmol/L正反引物各1µL、2.5 mmol/L dNTPs 4µL、5 U/µL LA Taq 0.3µL、2* GC PCR Buffer 12.5µL,去离子水补足体系至25 µL。反应条件设为:94℃预变性5 min、94℃变性1 min、67℃退火40 s、72℃延伸1 min,循环35次,最后72℃延长10 min。PCR产物经1.5%琼脂糖凝胶电泳鉴定后,送往生物公司测序。
1.2.2 TA克隆 为分离突变序列,将先证者PCR产物进行割胶纯化,将纯化的DNA凝胶回收产物4.5ul、pMD18-T Vector 0.5ul、DNA ligation Kit 5ul轻轻混合后,于4℃连接过夜,将DNA-质粒连接产物10µL与DH5α感受态细胞25µL相混合,在冰中放置30 min,然后经42℃热激反应45 s后,在冰中放置2 min,将上述反应液加入已预先37℃保温的SOC培养基,使其终体积为1 mL,37℃震荡培养1 h,然后取适量菌液均匀涂于LB培养基,37℃过夜静置培养,第2天挑选菌落后,加入到5 mL LB+Amp培养基中,过夜震荡培养,使用TIANprep Mini Plasmid Kit纯化质粒DNA,进行限制性内切酶酶切处理和琼脂糖凝胶电泳确认后,送往生物公司测序。
2 结果
2.1 临床表现该家系目前共有直系成员38人(配偶除外),根据基因检测结果,家系成员中连续3代均有罹患OPMD患者,具有显性遗传特点。但由于该病临床症状具有晚发性特点,目前确定出现症状者仅见于第Ⅰ代和第Ⅱ代患病家族成员。Ⅱ11和Ⅱ15目前年龄均已超过60周岁,并未出现任何肌病症状,结合该家系发病特点及基因检测结果,已基本确定两人并未存在PABPN1基因突变。现已发病并出现明确症状患者共有7例(见表1),其中男性5例,女性2例,发病年龄均约为50岁左右,发病患者中男性患者均以眼睑下垂为首发症状起病,而女性患者则以吞咽困难为首发症状,肌肉受累均以四肢近端为主,患者随年龄增大均有临床症状加重及不同程度讲话带鼻音和构音欠清情况。
2.2 基因检测正常多聚腺苷酸结合蛋白核1基因(PABPNl)的第1外显子上有一段正常的碱基序列为(GCG)6,测序峰图以单峰呈现(见图2-A),而参与基因检测的家族成员中,包括先证者在内共有10位存在PABPN1基因第一外显子中出现了(GCG)的异常重复扩增,碱基序列由(GCG)6突变为(GCG)10,测序峰图表现为突变点后出现双峰(见图2-B)。通过TA克隆,我们清晰地将突变序列分离出来(见图2-C)。
3 讨论

表1 OPMD已发病患者的临床资料
眼咽型肌营养不良(OPMD)起病隐匿,发病绝大多数具有显性遗传特点,极个别为隐性遗传,也有散发病案报道[6-7]。临床症状以颅神经支配肌和四肢近端肌群受累明显,主要表现为眼睑下垂,吞咽困难,构音不良以及四肢肌无力等症状,颜面肌受累可出现颜面肌的萎缩,通常心肌、平滑肌不受累[8]。绝大多数患者40岁以后开始出现症状,少数纯合子突变病例发病年龄可提前[9]。首发症状以眼睑下垂和吞咽困难为多见,但荷兰也有报道OPMD患者以肢体无力为首发症状起病的[10]。在我们所确诊的家系中,患者发病年龄均为50岁左右,男性患者均以眼睑下垂为首发症状,5~10年后开始出现四肢近端肌肉乏力,行走困难等症状,往后开始逐渐出现讲话带鼻音,构音欠清以及吞咽困难等表现。女性患者中目前确定出现症状者仅有2例,两人均以吞咽困难为首发症状,而后才开始出现眼睑下垂及行走困难症状,其中之一为先证者母亲,其在80余岁时因进食困难及反复肺炎发作离世,另外1例女性患者今年72岁,目前仅可进食软细食物,尚可借助步行辅助器行走。该患者家系中不同性别患者分别以不同症状起病,目前在世界各地所报道病例中,尚无相关统计数据论证性别与起病症状的相关性,在我们所确诊的家系中,由于出现明确症状患者较少(n=7),样本量尚不足以支持揭示该家系患者的起病方式与性别之间的关系,仍有待追踪观察家系中其他未发病患者的临床表现,但结合目前所报道的病例资料[11],携带(GCG)10的男性患者极有可能是以眼睑下垂为首发症状。

图2 家系先证者PABPN1基因异常扩增(GCG)的核苷酸链 A:为正常人PABPN1基因序列(GCG)6;B:为先证者PABPN1基因突变序列;C:为先证者PABPN1基因经克隆分离出的突变序列(GCG)10
该家系已发病患者中体查均可见双侧眼睑下垂,眼球活动障碍,以上视受限明显,无复视。患者讲话均带有明显鼻音,日常生活易以后仰头位弥补眼睑下垂带来的视野受限,即所谓的“占星师”姿势[12],以致于患者额纹较正常人明显加深(见图3),这也可看成是上睑下垂后的一种代偿机制,但Swart则认为这样的头部的扭曲也在一定程度上也加重了吞咽困难的症状[13]。OPMD患者晚期由于咽喉肌受累,吞咽及呼吸均受影响,容易导致反复吸入性肺炎及严重营养不良,这往往也是导致患者死亡的重要原因。

图3 OPMD家系中已发病患者不同年龄时期眼睑及额纹变化
OPMD自1998年确定致病基因位于14q11.2-13的多聚腺苷酸结合蛋白核1基因(PABPNl)的第1外显子上后,全世界各地均有OPMD病例的报道,而其中(GCG)9是最常见的突变类型,约占报告家系的69%[4]。我们所确诊的家系突变类型为(GCG)10,在目前所报告病例中相对少见。不同于Huntington病、齿状核红核苍白球路易体萎缩症(dentatoru⁃bral-pallidoluysian atrophy,DRPLA)等 长 序 列(GCG)重复扩增,发生在OPMD中的(GCG)扩充很温和,最高只加入7个(GCG)[4],并且在遗传上比较稳定,不存在随着世代遗传(GCG)n重复扩增次数增加,发病年龄年轻化,临床症状加重的遗传递进现象[11]。由于(GCG)编码为丙氨酸,OPMD患者的这些基因突变导致PABPN1蛋白末端形成了多个丙氨酸,这也被认为可能是PABPN1基因突变的一种致病方式,拥有了扩充的多丙氨酸序列的PABPN1可瞬间表达导致在核内聚集,进而伴随着细胞凋亡[14],这也是为什么用显微镜检查患者的肌肉可见规则的细丝状的蛋白凝块。不过遗憾的是,该家系患者因为年龄较大,均拒绝了接受肌肉活检,我们无法确定该家系患者肌肉病理方面的改变。虽然尚无证据表明(GCG)的拷贝数与临床表现有绝对相关性,但也有相关研究认为(GCG)9重复序列的患者症状会比其他突变类型重[15]。
虽然经过10余年的探索,不同国家均有相关的病例报告,但是OPMD的发病机制至今仍不清楚,对于OPMD的治疗也没有任何特效的方法。而动物模型的成功建立,已经为该病的临床治疗提供了可能。Davies等[16]利用OPMD转基因鼠研究发现,在疾病的开始阶段使用强力霉素,通过降低核内蛋白的聚集,可减轻转基因鼠的症状。但目前对于OPMD的药物治疗,仍然处于探索和实验阶段,过渡到临床应用还有很长的一段路要走。对于患者出现的临床症状,现有的治疗方案只能对症处理以及在早期加强肢体锻炼和康复理疗等措施,在一定程度上可起到延缓症状进展的作用,必要时可考虑外科手术治疗眼睑下垂及吞咽困难等症状。总的来说,该病至今尚无有效的治疗手段,做好产前检查以及遗传咨询,于产前明确诊断是预防本病的关键。
[1]Abu-Baker A,Rouleau GA.Oculopharyngeal muscular dystro⁃phy:recent advances in the understanding of the molecular pathogenic mechanisms and treatment strategies[J].Biochim Bio⁃phys Acta,2007,1772(2):173-185.
[2]Brais B.Oculopharyngeal muscular dystrophy:a polyalanine my⁃opathy[J].Curr Neurol Neurosci Rep,2009,9(1):76-82.
[3]Calado A,Carmo-Fonseca M.Localization of poly(A)-binding protein 2(PABP2)in nuclear speckles is independent of import into the nucleus and requires binding to poly(A)RNA[J].J Cell Sci,2000,113(Pt 12):2309-2318.
[4]Brais B,Bouchard JP,Xie YG,et al.Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy[J]. Nat Genet,1998,18(2):164-167.
[5]Robinson DO,Hammans SR,Read SP,et al.Oculopharyngeal muscular dystrophy(OPMD):analysis of the PABPN1 gene ex⁃pansion sequence in 86 patients reveals 13 different expansion types and further evidence for unequal recombination as the mu⁃tational mechanism[J].Hum Genet,2005,116(4):267-271.
[6]Gurtler N,Plasilova M,Podvinec M,et al.A de novo PABPN1 germline mutation in a patient with oculopharyngeal muscular dystrophy[J].Laryngoscope,2006,116(1):111-114.
[7]Tremolizzo L,Galbussera A,Tagliabue E,et al.An apparently sporadic case of oculopharyngeal muscular dystrophy:the first Italian report[J].Neurol Sci,2007,28(6):339-341.
[8]Abu-Baker A,Rouleau GA.Oculopharyngeal muscular dystro⁃phy:recent advances in the understanding of the molecular pathogenic mechanisms and treatment strategies[J].Biochim Bio⁃phys Acta,2007,1772(2):173-185.
[9]Brais B.Oculopharyngeal muscular dystrophy:a late-onset poly⁃alanine disease[J].Cytogenet Genome Res,2003,100(1-4): 252-260.
[10]Van Der Sluijs BM,Hoefsloot LH,Padberg GW,et al.Oculopha⁃ryngeal muscular dystrophy with limb girdle weakness as major complaint[J].J Neurol,2003,250(11):1307-1312.
[11]Muller T,Schroder R,Zierz S.GCG repeats and phenotype in oculopharyngeal muscular dystrophy[J].Muscle Nerve,2001,24 (1):120-122.
[12]Becher MW,Morrison L,Davis LE,et al.Oculopharyngeal mus⁃cular dystrophy in Hispanic New Mexicans[J].JAMA,2001,286 (19):2437-2440.
[13]de Swart BJ,van der Sluijs BM,Vos AM,et al.Ptosis aggra⁃vates dysphagia in oculopharyngeal muscular dystrophy[J].J Neurol Neurosurg Psychiatry,2006,77(2):266-268.
[14]Fan X,Dion P,Laganiere J,et al.Oligomerization of polyala⁃nine expanded PABPN1 facilitates nuclear protein aggregation that is associated with cell death[J].Hum Mol Genet,2001,10 (21):2341-2351.
[15]Pou Serradell A,Lloreta Trull J,Corominas Torres JM,et al.Oc⁃ulopharyngeal muscular dystrophy:study of patients from seven Spanish families with different GCG expansions in PABP2 gene [J].Neurologia,2004,19(5):239-247.
[16]Davies JE,Wang L,Garcia-Oroz L,et al.Doxycycline attenu⁃ates and delays toxicity of the oculopharyngeal muscular dystro⁃phy mutation in transgenic mice[J].Nat Med,2005,11(6): 672-677.
Clinical and molecular genetic studies of a Chinese family with oculopharyngeal muscular dystrophy.
CHEN Yonghong,LONG Yuesheng,CAI lili,WANG Hailong,MA Biao,FU Junyi,XIA Yong,LI Xinyi,XIE Longchang. Department of Neurology,the Second Affiliated Hospital of GuangZhou Medical University,GuangZhou 510260,China,Tel:020-34153276.
ObjectiveTo investigate the clinical and molecular genetic changes in a Chinese family with oculopha⁃ryngeal muscular dystrophy(OPMD).MethodsWe collected the clinical data of the familial members and blood sam⁃ples from all available 16 familial members,including the proband.The samples were analyzed using modified poly⁃merase chain reaction amplification and direct sequence analysis.ResultsMale OPMD patients initially presented with ptosis,followed by pronunciation difficulty,dysphagia and limb weakness whereas female OPMD patients initially pre⁃sented with swallowing difficulty.Genetic test revealed the abnormal expansions of the GCG trinucleotide repeat from GCG6 to GCG10 in PABPN1 gene in 10 familial members.ConclusionsThe genetic test and prenatal diagnosis is the key for the prevention treatment of oculopharyngeal muscular dystrophy.The ptosis of eyelid may be the initial symptom for the male patients of oculopharyngeal muscular dystrophy with(GCG)10mutation.
Ptosis Dysphagia Oculopharyngeal muscular dystrophy Polyadenylate binding protein nuclear 1 (PABPN1)gene
R746
A
2014-12-24)
(责任编辑:李立)
10.3936/j.issn.1002-0152.2015.06.004
☆ 广东省人口和计划生育委员会科研项目(编号:20110259)
* 广州医科大学附属第二医院神经内科(广州 510260)
△ 山西大医院神经内科
猜你喜欢
临床输血与检验(2022年3期)2022-06-22 02:52:50
阅读(科学探秘)(2021年12期)2021-05-30 10:11:06
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
中国医疗美容(2015年2期)2015-07-19 10:11:59
中国医疗美容(2015年2期)2015-07-19 10:11:59
中国医疗美容(2015年1期)2015-07-12 10:06:39
