
非布司他原料2种含量测定方法的比较研究
2015-01-17 08:39:18沈卫阳郭静沫沈圃毅金祥飞中国药科大学理学院南京210009药物质量与安全预警教育部重点实验室南京210009南京师范大学附属中学南京21000
西北药学杂志 2015年2期
沈卫阳,郭静沫,施 睿,沈圃毅,金祥飞(1.中国药科大学理学院,南京 210009;2.药物质量与安全预警教育部重点实验室,南京 210009;.南京师范大学附属中学,南京 21000)
·药物分析·
非布司他原料2种含量测定方法的比较研究
沈卫阳1,2*,郭静沫1,2,施 睿3,沈圃毅3,金祥飞1,2(1.中国药科大学理学院,南京 210009;2.药物质量与安全预警教育部重点实验室,南京 210009;3.南京师范大学附属中学,南京 210003)
目的 建立并比较滴定法和高效液相色谱法测定原料中非布司他含量。方法 化学分析法,用乙醇作为溶剂,氢氧化钠溶液为标准溶液,酚酞为指示剂进行滴定。高效液相色谱法,采用Inertsil ODS-SP(250mm×4.6mm,5μm)色谱柱;流动相为1 mL·L-1磷酸溶液(三乙胺调pH值至2.4)-乙腈(45∶55);流速为1.5mL·min-1;柱温为30℃;检测波长为315nm。结果 2种方法均符合方法学验证要求,测定同一样品,测量值差异无统计学意义(P<0.05)。结论 2种分析方法均能快速、有效地测定原料中非布司他的含量,均可作为非布司他原料质量控制的分析方法。
非布司他;含量测定;容量分析;高效液相色谱法
非布司他是一种非嘌呤、非竞争性的黄嘌呤氧化酶(XO)抑制剂[1],用于治疗痛风,2009年2月获美国食品药品管理局(FDA)批准,同年3月在美国上市。该药2010年获欧盟批准,先后在法国、英国和德国等多个国家上市[2],在中国于2013年2月上市[3]。与别嘌醇和安慰剂相比,非布司他在降低患者血尿酸水平方面更有效。轻中度肝功能损害及轻中度肾功能损害对非布司他的药动学参数无显著影响[4]。本文建立了酸碱滴定法和高效液相色谱法对非布司他原料的含量进行了测定,并对2种方法进行了比较[4-6]。
1 仪器与试药
1.1 仪器 LC-2010AHT高效液相色谱仪(包括LC-10AT泵、LC-20ADAD检测器和LC solution色谱工作站,日本岛津公司);UV-1800紫外分光光度仪(日本岛津);BS124S电子天平(赛多利斯科学仪器有限公司)。
1.2 试药 非布司他(批号20110612,20110616,20110620,由福州辰星药业有限公司提供);非布司他对照品(批号20110529,质量分数99.8%,由福州辰星药业有限公司提供);乙腈(色谱纯,Merck公司);超纯水(优普超纯水制造仪);其余试剂除注明外均为分析纯。
2 方法与结果
2.1 滴定法
2.1.1 滴定方法选择 非布司他在乙醇中略溶,在水中几乎不溶。从结构式(见图1)可以看出,非布司他中含有一个羧基官能团,可与碱发生酸碱中和反应,故选择乙醇作为溶剂,氢氧化钠为滴定液进行滴定。
取本品约0.65g,精密称定,加中性乙醇50mL溶解后,加酚酞指示液3滴,用氢氧化钠滴定液(0.1 mol·L-1)滴定。每1mL氢氧化钠滴定液(0.1 mol·L-1)相当于31.64mg的C16H16N2O3S。
2.1.2 滴定曲线的绘制与指示剂的选择 在滴定分析中,我们用电位滴定法进行了滴定终点的判断,并考察了酚酞作为指示剂,化学计量点前后溶液颜色的变化情况。滴定曲线如图2所示。

图1 非布司他的结构式Fig.1 Chemical structure of febuxostat

图2 滴定曲线Fig.2 Titration curve
由图2可知,该法滴定突跃大,终点颜色变化明显(从无色→淡红色),指示剂法判断滴定终点与电位滴定二阶微商线性内插法结果相一致。
2.1.3 成比例的系统误差、额外的系统误差与精密度实验 称取7份不同质量的非布司他供试品(0.163 4,0.337 9,0.415 3,0.495 9,0.571 2,0.653 5和0.730 8g),加中性乙醇50mL溶解后,加酚酞指示液3滴,用氢氧化钠滴定液(0.1mol·L-1)滴定,使滴定终点消耗的滴定液体积为所用滴定管体积(25mL)的20%,40%,50%,60%,70%,80%和90%,记录供试品的滴定体积。以滴定体积为因变量对供试品质量进行回归。实验结果见表1。

表1 滴定的线性方程Tab.1 Linear equation of titration
2.1.3.1 成比例的系统误差 按公式(1)和(2)进行计算。
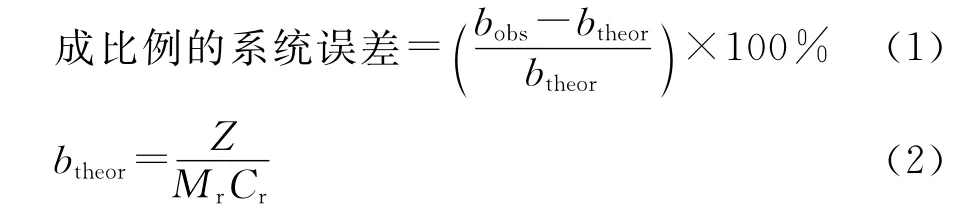
bobs为表1中线性方程计算得到的斜率;btheor为滴定常数;Mr为相对分子质量;Z为化学反应的计量系数;Cr为滴定液的浓度,本实验Cr=0.099 06 mol·L-1。
结果表明,成比例的系统误差为-0.06%,满足定量分析法中含量测定对成比例系统误差的绝对值小于0.5%的要求。
2.1.3.2 额外的系统误差 按公式(3)和(4)进行计算。

aobs为线性方程的截距;mT为供试品的预期称样量,本实验mT=0.65g;VT为预期或目标滴定体积。
结果表明,额外的系统误差为-0.32%,满足绝对值小于0.6%的要求。
2.1.3.3 精密度(统计学误差) 按公式(5)和(6)进行计算。
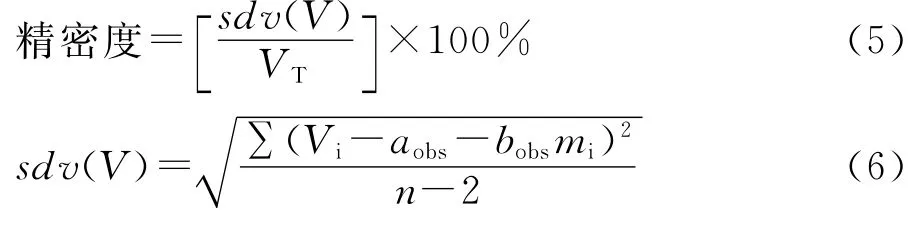
sdv(V)是标准偏差的估计值;Vi为消耗滴定液的体积;mi为供试品的实际称样量;n为滴定的次数。
结果表明,该方法的精密度为0.24%,满足小于0.5%的要求。
2.1.4 重复性实验 精密称取本品(批号20110612)6份,按2.1.1项下的方法进行操作,测定其含量,结果含量平均值为99.58%,RSD为0.12%,Δx值为-0.42%,符合含量限度99%~101%、重复性RSD<0.33%、相对准确度±0.67%的要求[7],因此,用酸碱滴定法测定非布司他含量是可行的。
2.2 高效液相色谱法
2.2.1 色谱条件 采用Inertsil ODS-SP(250mm ×4.6mm,5μm)色谱柱;流动相为乙腈-1mL·L-1磷酸溶液(三乙胺调pH值至2.4)(55∶45);检测波长为315nm;流速为1.5mL·min-1;进样体积20 μL;柱温为30℃。
2.2.2 对照品溶液及供试品溶液的配制 精密称取非布司他自制对照品约3mg,置于10mL量瓶中,加乙腈溶解并用流动相稀释至刻度,摇匀,精密量取5mL,置于50mL量瓶中,用流动相稀释至刻度,摇匀,即得对照品溶液。精密称取非布司他供试品约15mg,置于50mL量瓶中,加乙腈溶解并用流动相稀释至刻度,摇匀,精密量取5mL,置于50mL量瓶中,用流动相稀释至刻度,摇匀,即得供试品溶液。
2.2.3 线性范围考察 精密称取非布司他自制对照品14.90mg,置于50mL量瓶中,用乙腈溶解并用流动相稀释至刻度,摇匀,即得质量浓度为300 μg·mL-1的非布司他标准储备液。分别吸取该标准储备液0.10,0.20,0.50,1.00,2.00和5.00mL,置于10mL量瓶中,加流动相稀释至刻度,摇匀,制成质量浓度分别为2.98,5.96,14.90,29.80,59.60和149.00μg·mL-1的非布司他标准系列溶液。精密量取各标准系列溶液20μL,注入高效液相色谱仪,以峰面积(Y)为纵坐标、非布司他质量浓度(X)为横坐标,绘制标准曲线,得回归方程:Y=65 016 X-2 176,r=0.999 9。结果表明,非布司他质量浓度在2.98~149.00μg·mL-1范围内,与峰面积具有良好的线性关系。
2.2.4 精密度实验 取标准系列溶液中14.90 μg·mL-1的对照品溶液,连续进样6次,记录峰面积,计算其RSD为0.032%;取同一批原料(批号20110612),由不同分析人员在不同日期,在2台高效液相色谱仪上进行含量测定,结果平均含量为99.50%,RSD为0.50%(n=8),可见该方法进样精密度和中间精密度均良好。
2.2.5 重复性实验 取同一批号原料(批号20110612),平行测定6次,外标法计算含量,结果平均含量为99.67%,RSD为0.52%(n=6),表明重复性良好。
2.2.6 检测限与定量限 取标准系列溶液中2.98 μg·mL-1的对照品溶液,用稀释法测其检测限(S/N=3)和定量限(S/N=10),实验结果表明,在本色谱系统条件下,检测限约为0.07ng,定量限约为0.2ng。
2.2.7 稳定性实验 取1份非布司他含量测定供试品溶液(批号20110612)于0,2,4,8和24h分别进样,测得峰面积RSD为0.053%(n=6),可见其在24h内稳定性较好,能够满足测定要求。
2.2.8 方法专属性 非布司他原料(批号20110612)经酸、碱、高温、氧化和光照破坏后,配制成供试品溶液,各取20μL注入高效液相色谱仪,记录色谱图至主成分保留时间的3倍。结果降解产生的杂质均能与非布司他完全分离,分离度(R)均>1.5;主峰峰纯度均为1.000,表明非布司他主峰中不包含其他杂质峰。以上实验结果表明,本法专属性强,可用于原料中非布司他的含量测定。色谱图见图3。

图3 高效液相色谱图A.未破坏样品;B.酸破坏样品;C.碱破坏样品;D.氧化破坏样品;E.高温破坏样品;F.强光破坏样品Fig.3 HPLC chromatogramsA.non-destruction sample;B.acid destruction sample;C.alkali destruction sample;D.oxidative destruction sample;E.high temperature destruction sample;F.light destruction sample
3 滴定法与高效液相色谱法的比较
取非布司他原料药(批号20110612)用滴定法和高效液相色谱法分别平行测定6次,并对结果进行F检验和t检验,结果见表2。

表2 2种含量测定方法结果比较Tab.2 The results determined by the two methods (n=6)
由此认为,这2种方法精密度不存在显著差异(P<0.05)。
4 讨论
4.1 检测波长的选择 非布司他紫外吸收谱在217和315nm处有吸收峰,考虑217nm处接近末端吸收,干扰较大,基线噪声大,不易平衡,且315nm处吸收大,干扰小,故高效液相色谱法选择315nm为检测波长。
4.2 流动相的选择 本实验分别考察了磷酸体系、甲酸体系和乙酸体系,结果磷酸体系基线稳定,与最近杂质分离度较好。非布司他结构中含有羧基官能团,应将流动相调节为酸性,使其呈游离状态,考察结果表明,pH从4.0降到2.4,峰形有明显改善,pH继续从2.4降低到2.0,峰形几乎没有变化,pH过低对色谱柱伤害较大,所以最终选定pH为2.4。
4.3 含量测定方法的选择 原料药的纯度要求高,限度要求严格,含量测定注重方法的准确性。酸碱滴定法为容量分析法,操作较简单,准确度较高,可以不用对照品,并且避免了使用贵重仪器[8],在有关物质检查同时控制的条件下,可以首先考虑用滴定法;高效液相色谱法是进行新药包括原料药研发与质量控制的理想方法,分离效率高,精确度高,检测灵敏度高,操作自动化,应用广泛[9-11]。此2种方法均能准确有效地检测原料中非布司他的含量,可以根据分析目的和实验室条件,选择适宜的方法。
[1] Mapa J B,Pillinger M H.New treatments for gout[J].Curr Opin Investig Drugs,2010,11(5):499-506.
[2] 袁毅.痛风药物治疗进展[J].世界临床药物,2010,31(8):504-506.
[3] 刘玉艳,李阅东,唐建飞,等.抗痛风新药非布司他的临床研究进展[J].中国新药杂志,2014,23(10):1103-1107.
[4] Mukthinuthalapati M A,Bandaru S P K,Bukkapatnam V,et al.Development and validation of a stability-indicating RP-HPLC method for the determination of febuxostat(a xanthine oxidase Inhibitor)[J].J Chromatogr Sci,2013,51(10):931-938.
[5] 张聪,王绍杰,麻荣丽,等.高效液相色谱法测定非布司他的含量及有关物质[J].沈阳药科大学学报,2010,27(8):648-651.
[6] 张晓燕,高永良.非布索坦片有关物质测定方法的建立[J].中国新药杂志,2011,20(22):2257-2261.
[7] European Directorate for the Quality of Medicines &Health Care.Technical Guide for the Elaboration of Monographs[M].6th Edition.Strasbourg:Council of Europe,2011:69-72.
[8] 商军,王蓓,蔡金华,等.容量滴定法的方法验证与认可[J].中国兽药杂志,2010,44(9):24-27.
[9] 刘放,吴小平.HPLC法测定辛伐他汀胶囊的含量和有关物质[J].西北药学杂志,2013,28(5):488-492.
[10] 孙甜,熊小密,喻乙珊.高效液相色谱法测定来托司坦的含量及有关物质[J].西北药学杂志,2013,28(2):151-153.
[11] 董琦鑫,朱家俊,郁韵秋,等.高效液相色谱法测定原料药中埃索美拉唑的含量[J].复旦学报:医学版,2014,41(1):118-120.
Comparitive study on two assay methods of febuxostat
SHEN Weiyang1,2*,GUO Jingmo1,2,SHI Rui3,SHEN Fuyi3,JIN Xiangfei1,2(1.School of Sciences of China Pharmaceutical University,Nanjing 210009,China;2.Key Laboratory of Drug Quality Control and Pharmacovigilance Ministry of Education,Nanjing 210009,China;3.the Affiliated Senior High School of Nanjing Normal University,Nanjing 210003,China)
Objective To establish and compare the titration method and high performance liquid chromatography(HPLC)method for determining the content of febuxostat.Methods For chemical analysis,ethanol was used as the solvent,sodium hydroxide as the volumetric solution and phenolphthalein as an indicator for titration.The HPLC was carried out on an Inertsil ODS-SP(250 mm×4.6mm,5μm)column;the mobile phase consisted of 1mL·L-1phosphoric acid solution(adjust pH value to 2.4by triethylamine)-acetonitrile(45∶55)with a flow rate of 1.5mL·min-1;the column temperature was set at 30℃;the detection wavelength was 315nm.Results The 2methods both met the requirements of methodology validation,and there was no significant difference between the results obtained from the titration method and HPLC method(P<0.05).Conclusion The two methods can detect febuxostat content effectively and suitable for the quality control of febuxostat.
febuxostat;content determination;volumetric titrations;HPLC
10.3969/j.issn.1004-2407.2015.02.009
R927.2
A
1004-2407(2015)02-0134-04
2014-09-10)
*通信作者:沈卫阳,男,博士,副教授
郭静沫,女,在读硕士研究生
猜你喜欢
医学概论(2022年4期)2022-04-24 14:59:22
云南化工(2021年11期)2022-01-12 06:06:18
食品安全导刊(2021年20期)2021-11-28 00:56:56
世界最新医学信息文摘(2021年12期)2021-06-09 08:36:58
中学生数理化(高中版.高考理化)(2020年3期)2020-05-30 12:26:34
家庭医学(下半月)(2019年9期)2019-10-12 08:03:50
北京航空航天大学学报(2017年4期)2017-11-23 05:48:25
中国惯性技术学报(2017年1期)2017-06-09 08:15:14
系统工程与电子技术(2016年7期)2016-08-21 13:58:58
河北地质(2016年2期)2016-03-20 13:52:04
