
基于LC/MS的代谢组学数据并行处理研究
2015-01-16 10:44孙海涛杨志强李葆红陈德展
质谱学报 2015年6期
孙海涛,杨志强,李葆红,陈德展
(1.山东师范大学信息技术管理处,山东 济南 250014;2.山东师范大学实验室与设备管理处,山东 济南 250014;3.山东师范大学化学化工与材料科学学院,山东 济南 250014)
代谢组学是关于生物系统代谢物组成及变化规律的科学,是系统生物学的重要组成部分[1]。核磁共振(NMR)、GC/MS、LC/MS是代谢组学研究中常用的3种分析方法。与GC/MS和NMR技术相比,LC/MS技术因具有普适性、高灵敏度和特异性,更适于分析难挥发或热稳定性差的代谢物,因此被广泛应用于疾病诊断、药物分析等领域,现已成为代谢组学研究的主流技术[2-4]。根据不同的研究目的,代谢组学研究策略可分为非靶向代谢组学和靶向代谢组学。其中,靶向代谢组学预先清楚代谢物的成分,不需要进行繁琐的生物信息学数据处理;而基于LC/MS的非靶向代谢组学一般不对样品中的代谢物做预先鉴定,只是按照既定的流程进行样品预处理、代谢物提取、LC/MS全扫描检测、数据预处理等,因此数据处理工作相对繁重[5]。在非靶向代谢组学实验中,UPLC/MS在带来较高的出峰能力(Zucker大鼠尿样分析,1 min即可得到1 000多个峰)[6]、较好的分辨率和灵敏度的同时,也产生了大量的需要处理的原始数据。文献[7-8]通过对国内外的代谢组学数据处理进行分析研究认为,高效、准确的数据处理工作是未来代谢组学发展的重要方向。
MZmine是芬兰Matej等[9]开发的一款开源免费的代谢数据处理软件,该软件能够完成基于LC/MS模式产生的原始数据处理、可视化和分析等任务,具有准确的数据处理能力,被广泛应用于代谢组学研究[10-11]。随着LC/MS技术的发展,一次实验的单个样本数据文件就有几百M甚至几G,MZmine在单计算节点上处理全部样本数据常常耗时多天。为此,加州大学的代谢组学研究人员通过增加单节点处理器数目来提高处理速度,发现相较于单核处理器,四核处理器处理同样数据的速度可以提高20%~30%,但是该方法在处理太大文件时的效果不佳[12]。在单计算节点数据处理速度提升受限的情况下,数据处理并行化是提高数据处理速度的重要手段:蛋白质分析软件X!Tandem并行化以后,在20个双核处理器的计算节点上处理同样的计算任务的速度提高了40倍[13];在基于LC/MS代谢数据多变量的分析阶段,Par等[14]采取数据降维和划分时间窗并行的方法提高数据处理速度,黎建辉等[15]也提出了基于MapReduce的并行化方法提高化合物的LC/MS鉴定效率。
数据预处理是代谢组学研究最复杂、最耗时的工作,为提高数据处理速度,本研究提出一种数据并行的预处理过程并行化方法,即原始数据分组后由多个安装了MZmine软件的计算节点分别处理。本工作将对并行方法的可行性和效率进行分析,提出依据组成成分的保留时间对原始数据分组,满足并行计算的可行性要求;按照谱峰分组,实现并行处理的负载均衡,使得并行时间最短。希望通过该并行方法,解决单计算节点数据处理慢的问题,有效加快海量代谢数据处理的速度。
1 研究的理论基础
并行处理是指同时使用多个计算节点解决问题。一个问题的并行化需要考虑两个方面:一是问题可并行,即计算任务能分解成多个部分同时执行;二是并行处理的负载均衡问题,即多个计算节点下解决问题的耗时要少于单个计算节点下的耗时[16]。从程序和算法设计的角度看,并行处理可分为任务并行和数据并行。其中,任务并行是将处理问题的方法并行化;数据并行是把数据分解成多个数据子集分别处理,比任务并行简单。本研究采用对LC/MS产生的原始数据分组并行处理的方法。
1.1 基于保留时间的数据分组
基于LC/MS进行代谢物分析,待测样品经色谱仪分离时,组分的保留时间(tR)常用来作为成分鉴定的依据[17-19]。色谱仪作为质谱分析的进样装置,在质谱分析前对化合物进行分离,混合物中各个化合物依据其保留时间依次进入质谱仪。质谱仪连续扫描采集数据,每一次扫描得到一帧质谱图,将一帧质谱图中所有的离子强度相加,得到对应扫描时间的一个总离子流强度;总离子流随时间变化的图谱是总离子流色谱图(TIC),以离子强度为纵坐标,时间为横坐标。TIC可视为该次分析的色谱图,即反映该混合物在色谱柱中分离后各组分浓度随时间的变化[20]。
从TIC可以发现,当总离子强度为零或低于某一个阈值时,此时无组分检出,因此,峰强可以作为组分有无的判定条件。如果成分A在TIC中对应的时间段为[t1,t2],则对于原始数据而言,依据t1和t2划分数据可以保证成分A数据的完整性。此外,数据预处理阶段的主要工作是识别混合物含有的各种成分,一种成分的鉴定是由其自身的保留时间和离子强度决定的,与其他成分的数据关系是松散耦合的,因此,保留时间可以作为不同成分数据划分的依据。通过上述分析,按照保留时间对数据分组能够满足数据并行的可行性条件。
1.2 基于谱峰的负载均衡
应用软件并行处理的目的是缩短执行时间,一个原始数据文件在未并行处理之前,所有的预处理过程都是由一个计算节点单独完成的,该过程耗时较长;并行化以后,数据文件被分成多组,交由多个计算节点同时完成,以此达到缩短计算时间的目的。由于一个并行处理的执行时间受限于运行最慢的部分,所以负载均衡一直是并行程序设计中的一个重要因素[21]。
负载均衡考虑的首要因素是需要处理的数据的特点以及所应用软件的运行方式。在基于数据并行的处理模式中,数据划分的效果和质量会影响并行处理的效率[22]。对代谢数据进行划分要考虑代谢数据预处理的特点。代谢数据预处理包括峰识别、重叠峰解析、峰对齐和归一化等[23]。数据处理在TIC不同时间段的复杂度不同,在有谱峰出现的时间段,需要进行大量的数据计算,耗时较长;在没有组分数据检出的时间段,计算耗时较短。根据代谢数据预处理的这一特点,按照谱峰进行数据分组来实现并行处理的负载均衡。
按照谱峰,而不是样品检测时间对数据平均分组实现负载均衡,是根据代谢物组成成分的性质不同。因样品经过色谱仪分离后进入质谱仪的时间并不相同,而离子检测器是以固定频率进行扫描,在某个时间段可能并无成分被检测到,所以数据在整个仪器运行时间的分布并不均衡。在定长的时间段内,谱峰的数目并不完全相同,因此在任务分解时不能按照样品检测时间平均分段来分组数据。小鼠血清样本的总离子流色谱图和三维色谱-质谱图示于图1,可以发现数据在全检测时间分布的不均衡性。
此外,各个计算节点的计算能力、I/O、图形处理能力等也是负载均衡考虑的因素。在本研究中,为了简化问题的复杂度,将参与计算的节点配置成完全相同,目的是消除由于配置不同导致的负载不均衡问题;数据在每个计算节点上由MZmine独立完成预处理任务,计算过程中不同节点不需要交换数据,这也消除了由于数据通信带来的负载均衡问题;此外,由于数据预处理工作是由同一软件完成的,消除了处理方法不同造成的负载均衡问题。
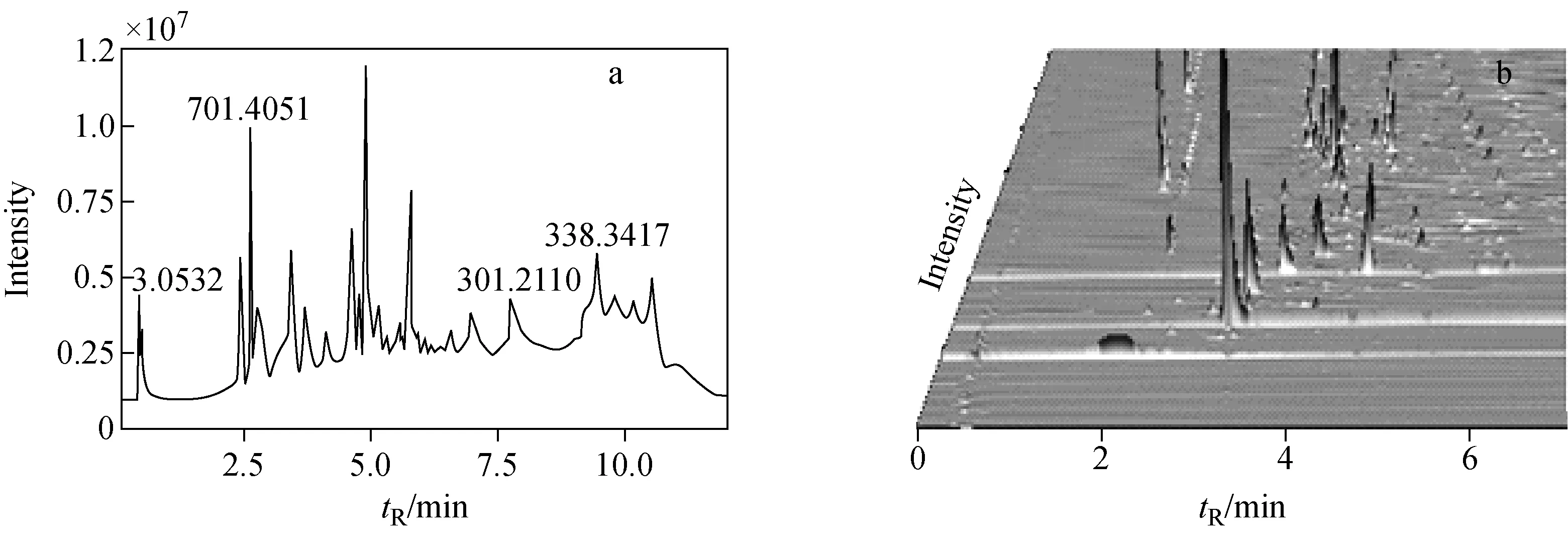
图1 小鼠血清样本的总离子流色谱图(a)和三维色谱-质谱图(b)Fig.1 TIC (a) and 3D chromatography mass spectrum (b) of the mice serum
1.3 谱峰的时间表示
按照谱峰分组数据的目的是提高并行效率,但是原始数据并不以谱峰形式存在。在TIC中,一个谱峰时间窗含有多个数据点(DP),一个数据点是一次仪器全扫描的结果。一般来讲,最窄的色谱峰至少包括10个DP,也有以20或40个点作为检测谱峰的标准[24-25]。以时间窗口表示谱峰,一个原始的TIC由多个时间窗组成,于是在按照谱峰进行负载均衡时,问题就变成了时间窗口的划分。在由分离完全的组分形成的原始谱图中,谱峰在时间轴上是一个时间窗[ts,te],ts表示一种组分经过色谱仪分离后开始进入质谱仪的时间,te表示这种组分从质谱仪完全流出的时间。当组分分离不完全时,在谱图上有重叠峰出现,虽然重叠峰是不同组分的集合,但是也可以表示为[ts,te]。在后续的谱峰预识别中,本研究将不再对峰和重叠峰进行区分,统一以峰来对待。
在谱峰预识别时允许存在重叠峰,是因为随着LC/MS技术的发展,特别是UPLC/MS的使用,多数组分能够得到完全的分离,表现在谱图上就是峰与峰之间有明显的边界,重叠峰在原始数据中只占很少的部分。在并行效率方面,谱峰的个数远远大于计算节点的个数,每个计算节点上实际分得大量的谱峰数据,这样即使由于少量重叠峰的存在导致某个计算节点在数据预处理阶段耗时长一点,但是相对单节点计算耗时(T串)以及并行处理时长(T并),额外耗时所占的比重也很小。在依据谱峰对数据分组并行时,忽略重叠峰不会对负载均衡造成较大的影响,但是却能明显降低数据分组的难度。
2 数据的并行处理
代谢数据依据保留时间分组满足了并行处理的可行性条件,按照谱峰分组能实现并行处理的负载均衡,谱峰的时间窗表示使得TIC中的谱峰与实测的按保留时间记录的数据实现了一一对应。代谢数据并行处理的流程是:管理节点接收原始数据后,按照样品检测时间对原始数据平均分组,分发给各个计算节点,计算节点对分组数据所包含的谱峰进行预识别;预识别完成后,管理节点对谱峰进行统计,再按照谱峰将原始数据平均分组,由MZmine完成代谢数据的预处理工作。为了便于陈述,在实际计算时以时间窗来划分谱峰数据,但在论述时仍然以按谱峰分组表示实现负载均衡的并行模式。
2.1 谱峰预识别
常用的谱峰识别方法有幅值法和斜率鉴别法。欧林军等[25]利用标尺与色谱曲线的交点来识别色谱峰;刘晓[26]利用迭代移动平均及归一化分析技术提高谱峰的识别率;这些方法在准确识别谱峰的同时也增加了计算的耗时。本研究提出了一种按照总离子强度对TIC中谱峰预识别的算法,该算法以i值确定色谱峰起始点和结束点,不对同一峰中的混合成分进行分离。谱峰预识别的目的是统计数据预处理总的工作量,不作为成分鉴定的依据。谱峰预识别算法分为谱峰时间窗识别和消除干扰值两个步骤。
2.1.1谱峰时间窗识别 原始的代谢数据由一些离散的点组成,每个点有一个数值对(t,i),这些离散的点构成了一个时间序列{(t1,i1),(t2,i2),…, (tR,in)}。在有谱峰存在的时间窗口i值呈现规律性的增加或减少,但都满足i>ib,ib是基线信号。设定x=i-ib,x是扣除基线信号后的强度,则峰与峰之间扫描点的x值为零,于是对谱峰的预识别变成寻找时间序列中连续的x非零的时间窗。
2.1.2消除干扰值 由于仪器或操作造成的误差,经过处理的数据仍然有很多x非零,但实际不是谱峰的时间窗,可以通过以下两种方法对这些时间窗进行排除:一是根据谱峰应包含的最少点的个数,连续的x>0点的个数多于20个的时间窗才能作为谱峰的候选;二是对多于20个点的时间窗,通过计算标准差排除非谱峰时间窗,只有在标准差大于一定值时才认为存在谱峰,即:
(1)
式中,N表示连续的非零点的个数,φ表示连续的偏离基线信号的噪音值。在实际操作中,φ需要经过不断调整才能既去除噪音,又保证不丢失谱峰信息。
通过上述算法完成对谱峰的预识别,得到记录谱峰的数组P,数组中每个元素记录了谱峰的起止时间,如Pn[tns,tne]中,tns表示第n个峰的开始时间,tne表示这个峰的结束时间。
2.2 谱峰预识别的并行化
谱峰预识别可以由单一计算节点完成,也可以并行处理。并行处理时,管理节点将原始数据接收进来后,按照样品检测时间对原始数据平均分组分发给每个计算节点。谱峰预识别并行处理数据分组示于图2(实线部分)。谱峰预识别算法的时间复杂度低,在并行处理时各个节点耗时相差不大,因此这一过程不考虑负载均衡。按照时间平均分组,在TIC上会出现谱峰被分割的情况,原始数据则是某段数据后面出现连续多个x大于0,但是总数又少于20个的点,在谱峰预识别时将这些点默认为一个谱峰,ts以第1个非0点开始的时刻为准。
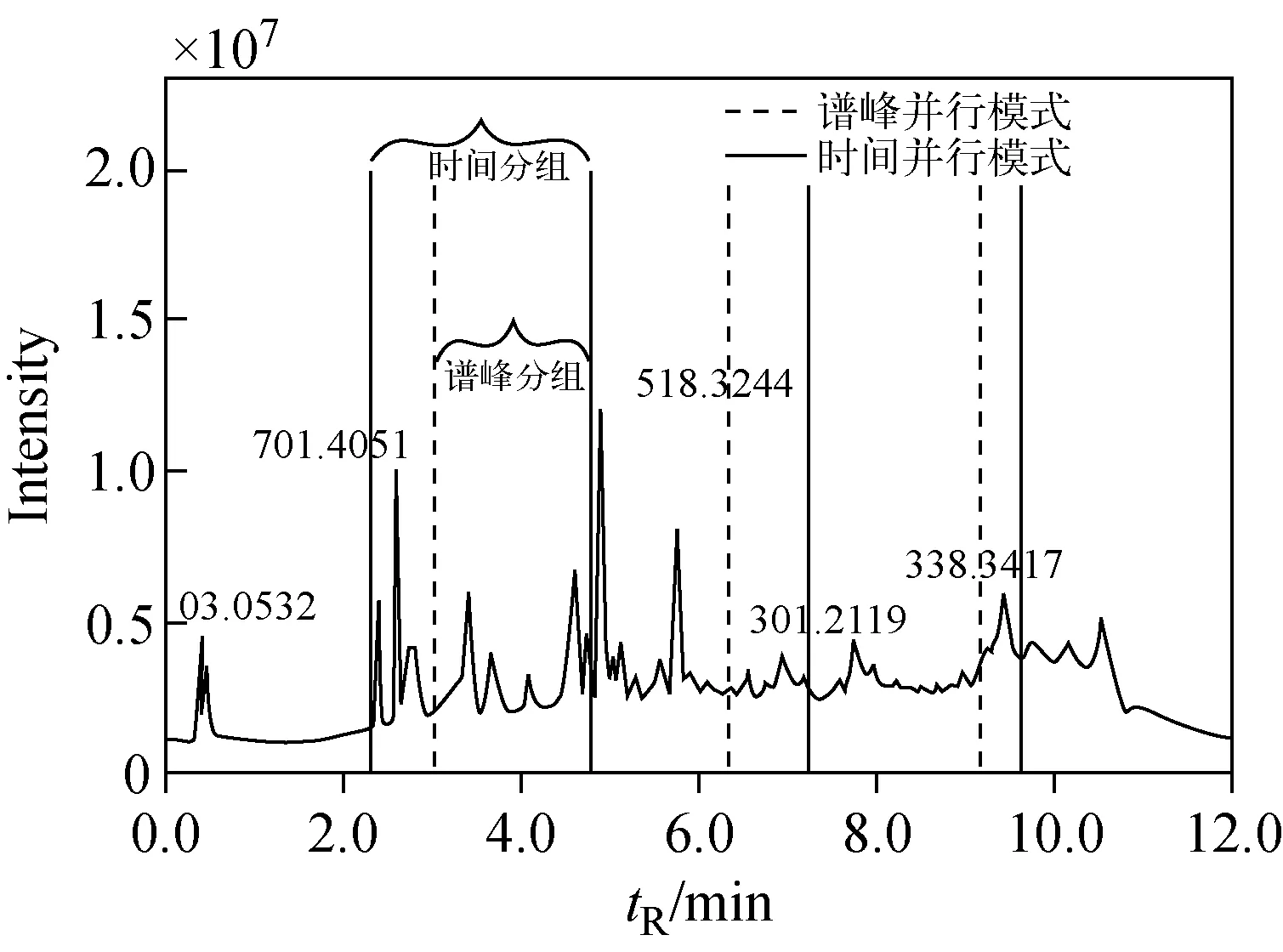
图2 原始数据不同分组方法示意图Fig.2 Different group methods for raw data
2.3 对数据预处理过程的并行化
谱峰预识别完成后,得到以谱峰为衡量的总工作量。按照谱峰对数据平均分组,就是将预识别的谱峰平均分配到每个计算节点上。如第m个节点分得的谱峰段为[Pl,Pk],则该节点实际分得数据的时间窗为[tls,tke],tls为第l个峰的开始时间,tke为第k个峰的结束时间,其分组方法示于图2(虚线部分)。
3 实验论证与分析
3.1 实验数据与并行环境
实验数据是小鼠血清样本经过UPLC-Q TOF-MS检测得到的,采用全扫描模式,样品检测时间为0~12 min。并行处理环境为5个配置完全相同的计算节点,1个为管理节点,4个为计算节点;管理节点在完成数据接收、分组、汇总等工作的同时也参与代谢数据预处理任务。5个节点都预装了MZmine和自行开发的代谢数据并行处理软件PMDP(parallel metabonomic data process)。PMDP具有完成代谢数据的接收、谱峰预识别、数据分组以及与MZmine通信等功能。
3.2 数据处理
实验数据处理分为3种模式:1) 单计算节点模式,由一个计算节点完成所有代谢数据预处理任务;2) 时间并行模式,按样品检测时间平均分段对数据分组的并行处理模式;3)谱峰并行模式,按谱峰对数据平均分组的并行处理模式。为了便于比较并行结果,本研究引入了相对时间(tr)的概念,即以单节点计算耗时(T串)作为基准时间,并行处理时,各计算节点耗时与之对比得到相对计算时间。如,单节点处理30个样品耗时为18 h,则T串=18,并行处理时某节点耗时为6 h,则该节点的相对时间tr=6/18=0.33。tr的引入是一种归一化处理方式,消除了样品本身性质的影响,从而使并行结果具有普遍意义。归一化后,t串=1,t并=Max(tr)。从图2可以看出,时间并行模式与谱峰并行模式两种分组方法在整个样品检测时间的数据分组不同;时间并行模式与谱峰预识别的数据分组方法相同。原始数据为27个血清样本,在单计算节点上预处理耗时约为23 h 40 min,2种并行处理模式的tr统计结果列于表1。

表1 同一数据2种并行模式的trTable 1 tr of two parallel computing modes
注:1)T是计算节点分组数据所在的时间窗,0~2.4 min是P1节点在按时间并行模式时,对这个时间窗口的数据进行预处理;
2) 在数据处理时,按照谱峰并行模式也是以时间窗来分组数据
3.3 实验结果分析
从表1可以看出:在并行时间的耗时方面,谱峰并行模式要少于时间并行模式;在负载效果方面,平均偏差大则说明各个计算节点的负载均衡不理想,因此,谱峰并行的负载均衡效果要更好一些。加速比(speedup)是指求解同一计算任务在单计算节点消耗的时间T串与在节点数为P的并行系统中消耗的时间T并的比值,即Sp=T串/T并,常用来衡量一个并行算法的效果[27]。加速比与本研究引入的相对时间的关系为Sp=1/tr。更多的实验数据证实,随着计算节点的增多以及代谢数据规模的扩大,谱峰并行模式的加速比Sp≈P;而时间并行模式的加速比则具有较大的随意性,在(1,P)之间波动,Sp与代谢物中成分组成有关。谱峰并行模式时,不同计算节点数目Sp趋势示于图3。
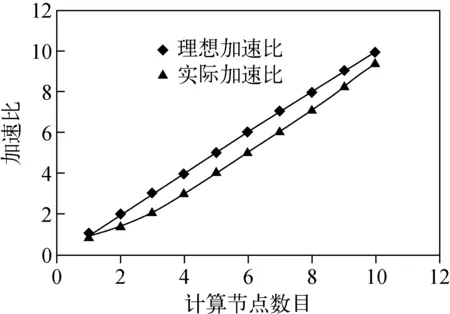
图3 谱峰并行模式时,不同节点数目的加速比Fig.3 Speedup of different node numbers in peak grouping mode
4 结论
本研究提出了一种基于LC/MS的代谢组学数据并行处理方法,原始数据分组后由成熟的代谢数据处理软件MZmine分别处理。实验结果表明,随着待处理数据的增多以及参与并行处理节点的增多,谱峰并行模式的加速比Sp趋近于线性加速比P。该方法部署简单、可扩展性强,可以解决单计算节点数据处理速度慢的问题,且能快速准确地处理基于LC/MS产生的海量数据。
[1] 王献,林树海,蔡宗苇. 基于质谱技术的代谢组学研究及其在中国的发展[J]. 中国科学:化学,2014,44(5):724-731.
WANG Xian, LIN Shuhai, CAI Zongwei. Mass spectrometry-based metabolomics and their developments in China[J]. Scientia Sinica Chimica, 2014, 44(5): 724-731(in Chinese).
[2] 李宁,范雪梅,王义明,等. 代谢组学及其分析技术的研究进展[J]. 中南药学,2014,12(7):668-673.
LI Ning, FAN Xuemei, WANG Yiming, et al. Development of metabolomics and its analytical technique[J]. Central South Pharmacy, 2014, 12(7): 668-673(in Chinese).
[3] 苏翠红,李笑天. 液相色谱和质谱联用技术及其在代谢组学中的应用[J]. 中华妇幼临床医学杂志:电子版,2010,6(1):62-64.
SU Cuihong, LI Xiaotian. High performance liquid chromatography mass sepctrometry and its application in metabonomics[J]. Chinese Journal of Obstetrics & Gynecology and Pediatrics: Electronic Edition, 2010, 6(1): 62-64(in Chinese).
[4] 王鹏远,张金兰. LC/MS技术在发现和鉴定药物中有关物质的应用[J]. 质谱学报,2010,31(6):362-367.
WANG Pengyuan, ZHANG Jinlan. Applications of LC/MS in discovery and characterization of related impurities in drug[J]. Journal of Chinese Mass Spectrometry Society, 2010, 31(6): 362-367(in Chinese).
[5] 赵春霞,许国旺. 基于液相色谱-质谱技术的代谢组学分析方法新进展[J]. 分析科学学报,2014,30(5):761-766.
ZHAO Chunxia, XU Guowang. Progress of metabonomics technique based on liquid chromatography-mass spectrometry[J]. Journal of Analytical Science, 2014, 30(5): 761-766(in Chinese).
[6] 谢跃生,潘桂湘,高秀梅,等. 高效液相色谱技术在代谢组学研究中的应用[J]. 分析化学,2006,34(11):1 644-1 648.
XIE Yuesheng, PAN Guixiang, GAO Xiumei, et al. Application of high performance liquid chromatographic technique in metabonomics studies[J]. Chinese Journal of Analytical Chemistry, 2006, 34(11): 1 644-1 648(in Chinese).
[7] 卢红梅,梁逸曾. 代谢组学分析技术及数据处理技术[J]. 分析测试学报,2008,27(3):325-332.
LU Hongmei, LIANG Yizeng. The development of analytical technologies and data mining in metabolomics[J]. Journal of Instrumental Analysis, 2008, 27(3): 325-332(in Chinese).
[8] 亓云鹏,胡杰伟,柴逸峰,等. 代谢组学数据处理研究的进展[J]. 计算机与应用化学,2008,25(9):1 139-1 142.
QI Yunpeng, HU Jiewei, CHAI Yifeng, et al. Advances of data analysis in metabonomics study[J]. Computers and Applied Chemistry, 2008, 25(9): 1 139-1 142(in Chinese).
[9] TOMAS P, SANDRA C, ALEJANDRO V B, et al. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data[J]. Bioinformatics, 2010, 11(1): 1-11.
[10] TREVINO V, YANEZ-GARZA L L, RODRIQUEZ-LOPEZ C E, et al. GridMass: A fast two-dimensional feature detection method for LC/MS[J]. Journal of Mass Spectrometry, 2015, 50(1): 165-174.
[11] COBLE J B, FRAGA C G. Comparative evaluation of preprocessing freeware on chromatography/mass spectrometry data for signature discovery[J]. Journal of Chromatography A, 2014, 1358: 155-164.
[12] TOBIAS K. West Coast Metabolomics Center at UC Davis. MZmine[EB/OL]. http:∥fiehnlab.ucdavis.edu/staff/kind/Metabolomics/Peak_Ali-gnment/mzmine/.
[13] DEXTER D, ANDREW L. Vanderbilt university school of medicine. Parallel Tandem[EB/OL]. http:∥www.thegpm.org/parallel/.
[14] PAR J, STEPHEN J B, THOMAS M, et al. Extraction, interpretation and validation of information for comparing samples in metabolic LC/MS data sets[J]. Analyst, 2005, 130(5): 701-707.
[15] 黎建辉,刘勇,王卫华,等. MapReduce计算模型下的化合物LC/MS鉴定[J]. 计算机科学与探索,2011,5(12):1 094-1 103.
LI Jianhui, LIU Yong, WANG Weihua, et al. LC/MS compounds identification under MapReduce[J]. Journal of Frontiers of Computer Science and Technology, 2011, 5(12): 1 094-1 103(in Chinese).
[16] 陈国良,孙广中,徐云,等. 并行计算的一体化研究现状与发展趋势[J]. 科学通报,2009,54(8):1 043-1 049.
CHEN Guoliang, SUN Guangzhong, XU Yun, et al. Integrated research of parallel computing: Status and future[J]. Chinese Science Bulletin, 2009, 54(8): 1 043-1 049(in Chinese).
[17] 潘芳芳. HPLC-QTOF-MS联用技术在药物杂质分析中的运用[D]. 杭州:浙江工业大学,2013.
[18] 张良晓. 气相色谱-质谱定性定量分析新方法研究[D]. 长沙:中南大学,2011.
[19] 邵晨,高友鹤. 色谱保留时间在蛋白质组研究中的应用[J]. 色谱,2010,28(2):128-134.
SHAO Chen, GAO Youhe. Application of peptide retention time in proteome research[J]. Chinese Journal of Chromatography, 2010, 28(2): 128-134(in Chinese).
[20] 蒋学慧. 色谱-质谱联用仪数据处理关键技术的研究[D]. 天津:天津大学,2013.
[21] 廖湘科. 网络并行计算中的负载平衡[J]. 小型微型计算机系统,1995,16(9):32-36.
LIAO Xiangke. Load balance in network parallel computing[J]. Mini-Micro Systems, 1995, 16(9): 32-36(in Chinese).
[22] 胡霞. 并行计算如何用于科学问题研究[J]. 科技资讯,2009:176.
HU Xia. Parallel computing in scientific research[J]. Science & Technologying Information, 2009: 176(in Chinese).
[23] 汪明明,程海婷,薛明. 基于LC/MS的代谢组学分析流程与技术方法[J]. 国际药学研究杂志,2011,38(2):130-136.
WANG Mingming, CHENG Haiting, XUE Ming. Recent development of LC-MS-based analytical procedures and techniques in metabonomics[J]. Journal of International Pharmaceutical Research, 2011, 38(2): 130-136(in Chinese).
[24] HANS J K, STAVROS K. 液相与气相色谱定量分析使用指南[M]. 陈小明,唐雅妍,译. 北京:人民卫生出版社,2010:43.
[25] 欧林军,曹建. 一种变压器油色谱峰识别算法的设计[J]. 色谱,2014,32(9):1 019-1 024.
OU Linjun, CAO Jian. A peak recognition algorithm designed for chromatographic peaks of transformer oil[J]. Chinese Journal of Chromatography, 2014, 32(9): 1 019-1 024(in Chinese).
[26] 刘晓. 识别色谱峰的一种方法[J]. 分析仪器,2005,(3):54-57.
LIU Xiao. A new method for distinguishing gas chromatographic peaks[J]. Analytical Instrumentation, 2005, (3): 54-57(in Chinese).
[27] 谢超,麦联叨,都志辉,等. 关于并行计算系统中加速比的研究与分析[J]. 计算机工程与应用,2003,39:66-68.
XIE Chao, MAI Liandao, DU Zhihui, et al. Research and analysis of parallel computing system speedup[J]. Computer Engineering and Applications, 2003, 39: 66-68(in Chinese).
猜你喜欢
昆明医科大学学报(2022年3期)2022-04-19
昆明医科大学学报(2021年4期)2021-07-23
分析科学学报(2021年3期)2021-07-14
色谱(2021年6期)2021-05-06
智慧健康(2021年33期)2021-03-16
物联网技术(2020年12期)2021-01-27
科技资讯(2020年12期)2020-06-03
空天防御(2020年1期)2020-04-13
天然产物研究与开发(2018年2期)2018-04-04
汽车零部件(2017年4期)2017-07-12
