
芳炔树脂/POSS固化相容性及热稳定性研究
2014-12-01 06:38范海波刘艳林杨荣杰李向梅
材料工程 2014年3期
范海波,刘艳林,杨荣杰,李向梅
(北京理工大学 材料学院 阻燃材料研究国家专业实验室,北京100081)
多面体低聚硅倍半氧烷(Polyhedral Oligomeric Silsesquioxane,简称POSS)是由Si与O组成内部无机骨架,外部连接有机基团的纳米级三维结构体系,分子为笼型结构,其有机取代基可为惰性有机基团或活性官 能 团[1,2]。由 于 POSS 的 几 何 尺 度 约 为 1~1.5nm,大小与高分子链段相当,其中,密度较大的无机内核能抑制聚合物分子的链运动而赋予杂化材料良好的耐热性能,相应的有机取代基则赋予杂化材料良好的韧性和可加工性,使POSS同时具备有机聚合物和无机陶瓷的基本特征,因此被称为新一代高性能聚合物材料[3]。
聚芳基乙炔(PAA)树脂是美国宇航航天局于20世纪80年代开发成功的一种全新的成炭树脂,其黏度低、高温残炭率高,适合用作炭/炭复合材料的浸渍剂;同时PAA也可作为聚合物复合材料的基体树脂,用于制造高温热结构材料和耐热烧蚀材料[4-6]。PAA树脂仅含有C、H元素,经炔基的加成固化后形成高度交联的芳环结构(图1),在惰性环境下加热至高温时,只有约10%的质量损失,残留率达85%~90%;其吸湿性仅有0.1%~0.2%,是酚醛树脂的1/50[7-9]。但PAA树脂也存在以下缺陷,固化放热量大且难以控制,极易发生爆聚现象;固化产物脆性大,易产生裂纹;树脂熔体与玻璃纤维或炭纤维的浸润性都比较差,造成复合材料易于分层、界面强度偏低。因此,PAA作为单纯的基体很难满足复合材料成型工艺要求[10,11]。

图1 PAA 树脂结构 (a)PAA 预聚物;(b)PAA树脂Fig.1 Molecular structure of PAA resin(a)prepolymer of PAA resin;(b)PAA resin
共混和共聚是制备新型复合材料的常用方法,在PAA中加入热稳定性优良的POSS分子可降低PAA体系中炔基的密度,从而降低其固化放热量,使复合体系固化过程易于控制;同时,POSS中硅元素的引入还有可能对PAA的热稳定性有一定的改善。由于PAA与惰性添加剂的相容性都较差,而POSS可通过对其分子的有机取代基团进行改性来提高PAA/POSS复合体系的相容性。笼型八苯基硅倍半氧烷[(C6H5SiO1.5)8,octaphenylsilsesquioxane,缩 写 为OPS]具有优异的热稳定性,常用于提高聚合物的热稳定性。本工作使用OPS及其衍生物笼型八氨基苯基硅 倍 半 氧 烷 (octa(aminophenyl)silsesquioxane,OAPS)和笼型八炔丙基胺苯基硅倍半氧烷(octa(propargylaminophenyl)silsesquioxane,OPAPS)对PAA树脂进行改性,研究了三种POSS与PAA树脂的相容性及热性能,POSS制备过程如图2所示。

图2 OAPS和OPAPS的制备Fig.2 Synthesis of OAPS and OPAPS
1 实验
1.1 仪器和试剂
树脂截面图由S4800型扫描电子显微镜测试得,对样品进行喷金处理,高真空条件下进行观察,C,O,Si元素的质量分数通过EDXS EX-350型X射线能谱仪测定;树脂热固化过程中的化学键变化由傅里叶变换红外仪(FTIR)测得,分辨率为4cm-1,扫描次数为32;树脂固化后由X射线衍射仪(XRD)测定其形态信息,Cu Kα射线源,扫描角度范围2θ=5~60°;树脂固化放热由差示扫描量热仪(DSC)测试,N2气氛下升温速率为10℃/min,升温范围30~360℃,N2流速20cm3/min;树脂固化后热稳定性由热重分析仪(TGA)测定,氮气和空气气氛,温度范围为40~900℃,升温速率为10℃/min。
PAA树脂:华东理工大学提供;笼型八苯基硅倍半氧烷:纯度97%,美国Hybrid Plastics公司;发烟硝酸、Na2CO3、乙醇、六水合三氯化铁、水合肼、乙酸乙酯、无水硫酸钠、正己烷、N,N-二甲基甲酰胺均为市售分析纯;四氢呋喃为市售色谱纯;5%Pd/C催化剂:宝鸡瑞科有限公司;80%丙炔溴甲苯溶液:天津Alfa Aesar有限公司。
1.2 笼型八氨基苯基硅倍半氧烷(OAPS)的制备
八氨基苯基硅倍半氧烷(OAPS)为自制,按照文献[12-14]由 OPS合成。OAPS:氢核磁(1H-NMR,500MHz,DMSO-d6,δ):7.4~6.2(2.0H,Ar H),5.4~4.5(1.0H,-NH2);固体 硅核 磁 (29Si solid NMR,400MHz,δ):-68.3,-77.5;碳 核 磁 (13C NMR,500MHz,DMSO-d6,δ):153.2,147.9,135.1,131.4,128.6,121.2,119.3,116.4,114.6,113.3;红外光谱(FT-IR,KBr,ν):3456and 3358(—NH2),1115(Si—O)cm-1;元素分析理论值:C 50.0,H 4.16,N 9.71;实测值:C 48.9,H 4.38,N 9.31。
1.3 笼型八炔丙基胺苯基硅倍半氧烷(OPAPS)的制备
八炔丙基胺苯基硅倍半氧烷(OPAPS)为自制,按照文献[15]由 OAPS合成。OPAPS:氢核磁(1H NMR,500MHz,DMSO-d6,δ):7.9~6.5(3.81H,Ar H),4.2~4.0(0.54H,—NH—),4.0~3.5(1.84H,—CH2—),3.1~2.8(1.00H,—C≡CH);固体硅核磁(29Si solid NMR,400MHz,δ):-77.9;碳核磁(13C NMR,500MHz,DMSO-d6,δ):162.4,146.9,128.9,124.3,120.0,79.5,75.1;红外光谱(FT-IR,KBr,ν):3288,2113and 642(—C≡CH),3375(—NH—),2922and 2827(—CH2—),1115(Si—O)cm-1;元素分析理论值:C 59.2,H 4.39,N 7.68;实测值:C 56.1,H 4.43,N 7.69。
1.4 POSS/PAA复合材料的制备
POSS粉末(0.4g)溶于少量沸点较低的有机溶剂中(OPS溶于二氯甲烷,OAPS或OPAPS溶于四氢呋喃),PAA 树脂预聚物(prePAA,1.6g)加入溶液,搅拌至均一。prePAA/POSS混合物在40℃下旋蒸除去溶剂,然后置于40℃的真空烘箱抽真空15min以抽除剩余的溶剂。prePAA/POSS混合物(2g)移到直径为3cm的称量瓶中置于真空烘箱中进行固化,固化程序为130℃/1h→150℃/1h→170℃/1h→190℃/1h→210℃/1h→230℃/1h→250℃/1h,得到厚度约为2~3mm的片状黑褐色固化产物。
2 结果讨论
2.1 PAA/POSS固化产物截面形貌特征
在prePAA中分别添加20%的OPS,OAPS和OPAPS后,可观察到固化产物截面有明显差异:PAA固化产物均一,外观为光亮黑色;加入OPS后发现固化产物分为明显的两层,上层为黑色,下层偏白,表明OPS未均匀分散,沉积在树脂下层;加入OAPS后,虽然最终固化产物外观均一,但在样品制备过程中,旋蒸除溶剂时可发现壁上有很多OAPS的颗粒存在;而加入OPAPS的样品,旋蒸除溶剂过程中壁上没有任何颗粒。因此,OPAPS与PAA相容性最好,OAPS次之,OPS最差。
图3为PAA和PAA/POSS固化产物截面SEM图。由图3中可看出PAA与PAA/20%OPAPS固化后产物截面光滑平整,分散均匀;而在PAA/20%OAPS固化产物的截面图中能看到很多颗粒存在。对图3中各图的不同位置进行能谱分析,结果如表1所示,由表1可得,PAA/20%OAPS固化产物截面白色颗粒区域Si元素含量远高于黑色光滑区域,表明白色颗粒区域为OAPS颗粒。因此,PAA与OAPS的固化过程中,OAPS仅为物理分散,二者不发生反应,且OAPS的颗粒直径大约为2μm;而PAA与OPAPS的固化过程中,OPAPS参与反应,形成均匀的固化产物。图4为PAA/OAPS和PAA/OPAPS固化产物可能的结构示意图。

图3 PAA和PAA/POSS固化产物截面SEM 图 (a)PAA;(b)PAA/20%OAPS;(c)PAA/20%OPAPSFig.3 SEM micrographs of PAA and PAA/POSS (a)PAA;(b)PAA/20%OAPS;(c)PAA/20%OPAPS
2.2 PAA/POSS固化产物XRD表征
图5为PAA/POSS固化产物的XRD衍射图。
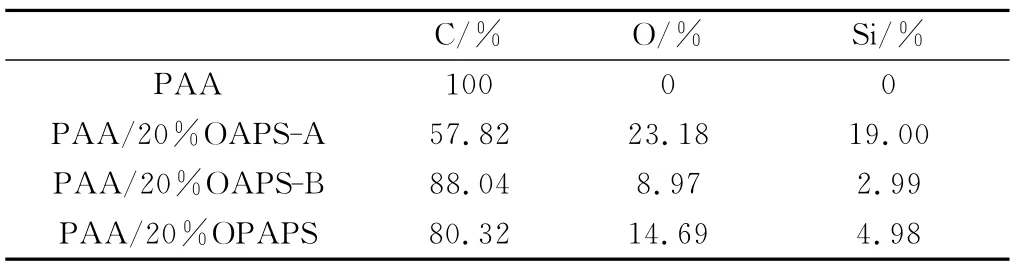
表1 PAA和PAA/POSS固化产物截面纶谱分析结果(质量分数)Table 1 EDXS results of PAA and PAA/POSS(mass fraction)

图4 PAA/OAPS和PAA/OPAPS复合物结构Fig.4 Structures of PAA/OAPS and PAA/OPAPS composites
从图5可看出,PAA固化产物为非晶态;PAA/20%OPS固化产物的下层产物中出现OPS很明显的衍射峰,而在上层的产物衍射图为PAA,证明OPS与PAA宏观上分相;PAA/20%OAPS固化产物XRD衍射图中在2θ=6.80°处出现一个小的衍射峰,此峰为OAPS分子尺寸的衍射峰,由此更可以证明OAPS未与PAA 发 生 反 应,仅 起 添 加 剂 的 作 用[16,17];PAA/20%OPAPS固化产物XRD衍射图显示其为非晶态,未见OPAPS分子尺寸的衍射峰,表明OPAPS分子中的端炔基与PAA的端炔基发生固化交联反应。

图5 PAA/POSS固化产物XRD衍射图Fig.5 X-ray diffraction patterns of PAA/POSS
2.3 红外表征
图6为prePAA,三种固化的PAA,PAA/20%OAPS,PAA/20%OPAPS产物的红外谱图对比。由红外谱图可知,PAA固化后,3288cm-1和610cm-1处的—C≡CH基团振动峰减小;同时,在PAA及PAA/POSS固化产物中,3024cm-1出现CAr—H键的伸缩振动峰,2963cm-1出现—CH=CH—基团上的C—H键的伸缩振动峰,1900cm-1出现芳香环C=C双键泛频的吸收峰,以及1594cm-1处出现C=C键的伸缩振动峰,表明PAA中的端炔基发生交联固化反应,生成共轭烯及芳环结构。与PAA固化后产物红外图相比,PAA/20%OAPS和 PAA/20%OPAPS固化后产物在1102cm-1与460cm-1处出现了Si-O键的吸收峰,表明POSS的笼型结构得以保留;且OAPS的伯胺(3383,3458cm-1)和 OPAPS的仲胺(3374cm-1)基团的吸收峰依然存在,表明伯胺或仲胺基团在与炔基的固化过程中不发生反应。

图6 PrePAA,PAA,PAA/20%OAPS和PAA/20%OPAPS红外谱图Fig.6 FT-IR spectra of uncured PAA and cured PAA/POSS composites
2.4 prePAA和prePAA/POSS热固化行为对比
图7是prePAA,OPAPS,prePAA/20%OAPS和prePAA/20%OPAPS在升温速率为10℃/min时的DSC曲线。在图7中,ΔH为固化放热量,Tp为固化放热峰峰值温度。从图中可看出,prePAA、prePAA/20%OAPS和prePAA/20%OPAPS三者放热峰峰形相似,位置稍有不同。prePAA/20%OAPS的放热峰位置与prePAA相比,基本没有发生变化,Tp也基本没有变化,表明OAPS与PAA不发生化学反应。而在prePAA中加入20%OPAPS之后,由于OPAPS与PAA之间发生共聚反应,树脂预聚物放热峰位置整体向高温区稍有偏移,Tp稍有增加,处于prePAA和OPAPS二者Tp之间;由于OPAPS放热温度更高,因此在225℃到300℃之间,prePAA/20%OPAPS放热量比prePAA或prePAA/20%OAPS体系的放热量更大。此外,在prePAA中加入POSS后,减小了体系中端炔基的密度,导致prePAA/POSS体系固化过程中放热量有所减小,加入POSS有助于固化反应的控制。
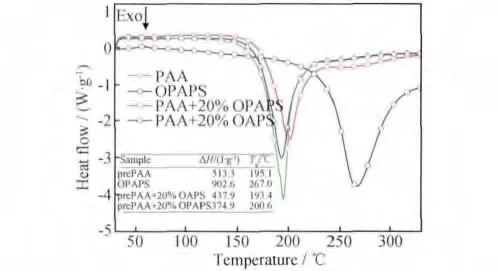
图7 PrePAA,OPAPS和prePAA/POSS的DSC曲线图Fig.7 DSC curves of prePAA,OPAPS and prePAA/POSS
2.5 PAA和PAA/POSS热稳定性
图8为PAA和PAA/POSS固化产物在N2和空气气氛下的TGA曲线。在图8中,Td5为失重5%时对应的温度,Tmax为失重速率最快时对应的温度,Residues为900℃时的残炭剩余百分数。由图可知,PAA固化产物在氮气气氛下,初始分解温度(Td5)很高,达到558℃,且它的残炭量也极高,达到了86.7%。加入OPAPS或者OAPS后,由于POSS的分解温度要低于PAA固化产物,故PAA/POSS固化产物的热稳定性稍有下降,分解温度下降约30~40℃。残炭率也稍有下降,但仍大于85%,比文献中用酚醛树脂等改性PAA的 热 稳 定 性 要 好[3,18]。在 空 气 气 氛 下,加 入POSS后,复合体系的热氧稳定性稍有提高,得益于加入POSS中Si元素的影响,体系Tmax提高了20℃左右,且残炭量也稍有提高。

图8 PAA和PAA/POSS的 TGA曲线 (a)氮气;(b)空气Fig.8 TGA curves of PAA,PAA/POSS (1)in N2;(2)in air
3 结论
(1)使用三种笼型硅倍半氧烷 OPS,OAPS和OPAPS对芳炔树脂PAA进行改性。在prePAA中加入20%的三种POSS后进行固化,发现OPS与PAA的相容性差,OPS沉积于PAA底部;OAPS与PAA的相容性好于OPS,但固化产物中仍能发现OAPS的颗粒存在;OPAPS的炔基可参与PAA的固化反应,故OPAPS与PAA的相容性最好。
(2)PAA和PAA/OPAPS固化反应后,炔基间反应生成共轭烯结构及芳环结构,固化产物中POSS笼型结构保留;且OAPS的伯胺和OPAPS的仲胺基团不参与树脂的固化反应。
(3)在PAA预聚物中加入OAPS或OPAPS后,体系端炔基的密度降低,固化过程中放热量有所减小,有助于固化反应的控制。PAA/OAPS体系固化放热峰形不发生变化;PAA/OPAPS体系固化放热峰向高温区偏移。PAA/POSS复合固化产物基本保持了PAA树脂的热稳定性。
[1]LICKISS P D,RATABOUL F.Fully condensed polyhedral oligo-silsesquioxanes(POSS):from synthesis to application[J].Adv Organomet Chem,2008,57:1-116.
[2]CORDES D B,LICKISS P D,RATABOUL F.Recent developments in the chemistry of cubic polyhedral oligosilsesquioxanes[J].Chem Rev,2010,110:2081-2173.
[3]杜建科,杨荣杰.笼形八苯基硅倍半氧烷的合成及表征[J].精细化工,2005,22(6):409-411.DU J K,YANG R J.Synthesis and characterization of polyhedral octaphenylsilsesquioxane[J].Fine Chemicals,2005,22(6):409-411.
[4]KATZMAN H A,MALLON J J,BARRY W T.Polyarylacetylene-matrix composites for solid rocket motor components[J].J Adv Mater,1995,26(3):21-27.
[5]ZHANG X Z,HUANG Y D,WANG T Y,etal.Influence of fibre surface oxidation-reduction followed by silsesquioxane coating treatment on interfacial mechanical properties of carbon fibre/polyarylacetylene composites[J].Compos Part A,2007,38(3):936-944.
[6]王明存,魏柳荷,赵彤,等.一种加成固化型热固性树脂PNPAA固化过程和热稳定性研究[J].高分子学报,2006,(2):241-246.WANG M C,WEI L H,ZHAO T,etal.Cure and thermal stability of an addition cure type thermoset PN-PAA[J].Acta Polymerica Sinica,2006,(2):241-246.
[7]ZALDIVAR R J,KOBAYASHI R W,RELLICK G S.Carborane-catalyzed graphitization in polyarylacetylene-derived carboncarbon composites[J].Carbon,1991,29(8):1145-1153.
[8]ZHANG X Z,HUANG Y D,WANG T Y,etal.Effects of polyhedral oligomeric silsesquioxane coatings on the interface and impact properties of carbon-fiber/polyarylacetylene composites[J].J App Polym Sci,2006,102(6):5202-5211.
[9]姚冬梅,程文,邹武,等.新型含硅芳基乙炔的固化及成炭机理研究[J].固体火箭技术,2009,32(5):578-582.YAO D M,CHENG W,ZOU W,etal.Study on curing and pyrolytic mechanism of new PSAA resin[J].Journal of Solid Rocket Technology,2009,32(5):578-582.
[10]WANG M C,ZHAO T.Polyarylacetylene blends with improved processability and high thermal stability[J].J App Polym Sci,2007,105(5):2939-2946.
[11]WANG S K,LI M,GU Y Z,etal.Experimental study on crack defects formation in polyarylacetylene composites and modification improvement of resin[J].Compos Mater,2010,44(25):3017-3032.
[12]范海波,杨荣杰,李定华.多面体低聚八氨基苯基硅倍半氧烷合成方法改进及其表征[J].化学学报,2012,70(4):429-435.FAN H B,YANG R J,LI D H.Synthesis improvement and characterization of polyhedral oligomeric octa(aminophenyl)silsesquioxane[J].Acta Chimica Sinica,2012,70(4):429-435.
[13]范海波,杨荣杰,李向梅.多面体低聚八硝基苯基硅倍半氧烷纯度分析[J].化学学报,2012,70(16):1737-1742.FAN H B,YANG R J,LI X M.Purity analysis of polyhedral oligomeric octa(nitrophenyl)silsesquioxane[J].Acta Chimica Sinica,2012,70(16):1737-1742.
[14]FAN H B,YANG R J.Synthesis and characterization of polyhedral oligomeric azido-cctaphenylsilsesquioxane[J].J App Polym Sci,2012,124(5):4389-4397.
[15]FAN H B,HE J Y,YANG R J.Synthesis,characterization,and thermal curing of a novel polyhedral oligomeric ccta(propargylaminophenyl)silsesquioxane[J].J App Polym Sci,2012,127(1):463-470
[16]LI Q,ZHOU Y,HUANG X D,etal.Synthesis and characterization of a novel arylacetylene oligomer containing POSS units in main chains[J].Eur Polym J,2008,44(8):2538-2544.
[17]ZHANG Z P,LIANG G Z,REN P G,etal.Curing behavior of epoxy/POSS/DDS hybrid systems[J].Polym Compos,2008,29(1):77-83.
[18]闫联生.酚醛改性聚芳基乙炔基复合材料探索[J].玻璃钢/复合材料,2001,(2):22-24.YAN L S.Property improvement of polyarylacetylene matrix composites by phenolic resins[J].FRP/CM,2001,(2):22-24.
猜你喜欢
农药科学与管理(2019年8期)2019-11-23
铜仁学院学报(2018年6期)2018-07-05
衡阳师范学院学报(2016年3期)2016-07-10
中国塑料(2016年7期)2016-04-16
华东理工大学学报(自然科学版)(2015年4期)2015-12-01
合成材料老化与应用(2015年4期)2015-07-25
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
化学工业与工程(2015年1期)2015-02-10
应用化工(2014年1期)2014-08-16
无机化学学报(2014年12期)2014-02-28
