
丙戊酸协同伊马替尼诱导K562细胞凋亡并下调Bcr/Abl mRNA和磷酸化蛋白激酶B的表达*
2014-11-08 02:27张祥忠朱小玉陈运贤
中国病理生理杂志 2014年1期
陆 英, 张祥忠, 丁 倩, 朱小玉, 陈运贤
(中山大学附属第三医院 1血液科,2输血科,3血液病研究所,广东 广州510630;4贵州省人民医院血液科,
贵州贵阳550002;5安徽省立医院血液科,安徽合肥230001;6中山大学附属第一医院血液科,广东广州510080)
慢性粒细胞白血病(chronic myelogenous leukemia,CML)是一种发生在造血干细胞的恶性骨髓增殖性疾病,其发病率为1/105~2/105。CML的特征性细胞遗传学改变为t(9;22)形成Ph染色体,由此形成的Bcr/Abl融合基因是其发病的分子机制。2001年美国FDA批准了第1个Bcr/Abl酪氨酸激酶抑制剂——伊马替尼(imatinib,STI571)治疗CML取得良好临床疗效,然而随着用药时间的累积,耐药成为了一个新问题。因此,开发新一代酪氨酸激酶抑制剂或者探求其它不同作用机制的抗肿瘤药物成为研究热点。近年来的研究表明组蛋白脱乙酰酶抑制剂(histone deacetylase inhibitors,HDACI)可诱导多种肿瘤细胞生长阻滞、分化及凋亡[1-2]。丙戊酸(valproate,VPA)是一种传统的抗癫痫药物,同时也表现出很强的 HDACI活性。本实验观察 VPA联合STI571对K562细胞增殖的抑制作用及其可能机制,为VPA应用于CML的治疗探索新的思路。
材料和方法
1 主要试剂
VPA购自法国赛诺菲安万特公司,STI571购自瑞士诺华公司。RPMI-1640培养基为Gibco产品,胎牛血清(fetal calf serum,FCS)购自杭州四季青生物公司,细胞周期分析试剂盒为 Becton Dickinson产品,Annexin/PI凋亡检测试剂盒购自碧云天生物技术研究所,Trizol购自Invitrogen,逆转录试剂盒购自Fermentas,Taq DNA聚合酶购自 Fermentas。PCR引物及探针由Invitrogen合成。抗蛋白激酶B(protein kinase B,PKB)单克隆抗体、抗磷酸化PKB(p-PKB)单克隆抗体以及抗磷酸甘油醛脱氢酶(glyceraldehyde phosphate dehydrogenase,GAPDH)单克隆抗体均购自Cell Signaling Technology,羊抗兔IgG和兔抗鼠IgG购自Santa Cruz。
2 细胞和细胞培养
K562细胞株由中山大学肿瘤研究所赠送。K562细胞株用含10%FCS、1×105U/L青霉素和100 mg/L链霉素的RPMI-1640培养基,在37℃、5%CO2及饱和湿度条件下培养,每隔2~3 d换液1次。
3 流式细胞术检测细胞周期
取对数生长期的K562细胞以5×109cells/L种于6孔板中,设立对照组、VPA组和 VPA联合STI571组,加药48 h后,收集各组细胞,磷酸盐缓冲液(phosphate-buffered solution,PBS)洗2次,离心弃上清,70%冷乙醇4℃固定过夜后,离心弃去固定液,3 mL PBS重悬5 min,400目筛网过滤1次,再离心5 min,弃去PBS。用1 mL PI染液染色,4℃避光30 min。上机检测,进行结果分析。
4 Annexin V-PI双染测定细胞凋亡
分别收集对照组、VPA组、STI571组和VPA联合STI571组的细胞,PBS洗2次,离心弃上清,悬于结合缓冲液中,调整细胞浓度为1×109cells/L。加10 μL FITC 标记的 Annexin V 和 10 μL PI,混匀,避光室温孵育10 min。用结合缓冲液洗1次后用CoulterElite流式细胞仪进行分析。Annexin V(-)PI(-)为活细胞,AnnexinV(-)PI(+)为机械损伤细胞,AnnexinV(+)PI(-)为早期凋亡细胞,AnnexinV(+)PI(+)为晚期凋亡细胞。
5R T-PCR检测mRNA水平
按照Trizol试剂说明书进行。收集各组细胞,PBS洗涤,离心弃上清后加入1 mL Trizol,15~30℃放置5 min。加入 200 μL氯仿,放置 2~3 min,13 000 r/min,4℃离心15 min。将上层水相层转到一新的离心管,加入异丙醇500 μL,放置10 min,13 000 r/min,4℃离心 10 min,弃上清。用 1 mL 75%乙醇洗涤1遍,空气中晾干。加入50 μL灭菌无RNA酶的超纯水溶解。取5 μL RNA溶液稀释100倍测 A260和 A280值,计算 A260/A280比值及最终RNA的含量。最后取5 μL RNA溶液用0.8%琼脂糖凝胶电泳鉴定。取2 μg RNA加入1 μL引物并以DEPC水配成终体积为25 μL的反应体系。将上述反应体系在70℃预变性5 min,立即放于冰上,加入下列物质:5 μL 5 × RT Buffer、1 μL 25 mmol dNTPs、25U RNasin、200 μmol/L M-MLV 逆转录酶,37 ℃1 h,95℃ 5 min反应后取得cDNA。
然后荧光定量PCR检测相关基因mRNA水平,反应体系为 10 μL 5 × PCR Buffer、15 pmol上下游引物、1 μL 10 mmol dNTPs、7.0 pmol探针、3U Taq DNA聚合酶和5 μL待测样本或阳性定量模板(cDNA),最后加无菌去离子水配成终体积为50 μL反应体系。相关基因的引物见表1。

表1 引物和探针序列Table 1.The primer and probe sequences
6 Western blotting检测蛋白水平
取对数生长期不同组别的K562细胞,PBS洗2遍,加蛋白抽提液抽提总蛋白,测定蛋白浓度,取蛋白50 μg,经10%十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)分离,常规转移至聚偏二氟乙烯膜(polyvinylidene fluoride,PVDF),用含5%脱脂牛奶的 TBST(Tris-buffered saline and Tween 20)封闭液封闭过夜,用Ⅰ抗4℃孵育过夜,1∶2 000稀释的辣根过氧化酶(horseradish peroxidase,HRP)标记的山羊抗小鼠Ⅱ抗室温孵育1 h,用 TBST洗3次,增强型化学发光试剂(enhanced chemiluminescent,ECL)显色液显色目的条带,用ImageJ分析软件进行灰度值分析,以GAPDH作为内参照。
7 统计学处理
数据用均数±标准差(mean±SD)描述。使用统计软件SPSS 13.0分析,两组以上均数比较采用单因素方差分析,方差齐时采用LSD检验进行组间两两比较,方差不齐时采用Tamhane检验进行组间两两比较,以P<0.05为差异有统计学意义。
结 果
1 VPA与STI571联合用药对K562细胞周期和细胞凋亡的影响
我们前期研究[3]获得了VPA处理K562细胞的浓度-细胞生长抑制率曲线以及时间-细胞生长抑制率曲线,根据结果,我们取 VPA的实验浓度为2 mmol/L。此浓度的 VPA 和0.5 μmol/L STI571联合处理K562细胞,与未处理组(对照组)相比,48 h后细胞周期发生阻滞,主要表现为G0/G1期细胞增多(79.73% ± 2.30%vs 40.13% ±2.12%),S 期细胞减少(18.19% ±1.32%vs 50.42% ±2.65%),差异均有统计学意义(P<0.05)。两药联合与单药作用相比,其阻滞作用无明显差异,而细胞凋亡则有明显增加(P <0.05),见图1、2 和表2。
2 VPA与STI571联合用药对K562细胞Bcr/Abl mRNA表达水平的影响
2 mmol/L VPA 和0.5 μmol/L STI571 联合处理K562细胞24 h后,VPA组以及联合用药组检测不到Bcr/Abl mRNA,而单用STI571组仍然可以检测到一定数量水平的 mRNA [(64.17±12.27)×109copies/(g total mRNA)],而且与对照组的差异有统计学意义(P <0.05),见表2。
3 VPA与STI571联合用药对K562细胞总PKB和p-PKB蛋白水平的影响
2 mmol/L VPA 和0.5 μmol/L STI571 联合处理K562细胞,加药后5、10、30 min以及24 h分别检测了细胞总PKB和p-PKB蛋白水平,结果发现在处理5 min后,单药组以及联合用药组的p-PKB蛋白水平均下降,直到24 h后几乎检测不到,而总PKB蛋白水平无明显变化,见图3、表3。
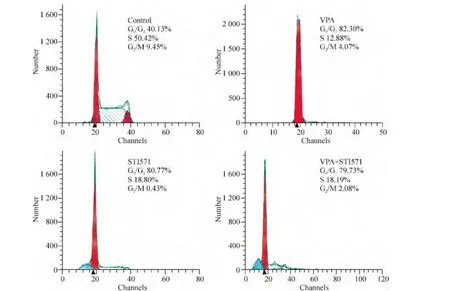
Figure 1.Effects of VPA and STI571 on the cell cycle of K562 cells.图1 VPA与STI571对K562细胞细胞周期的影响
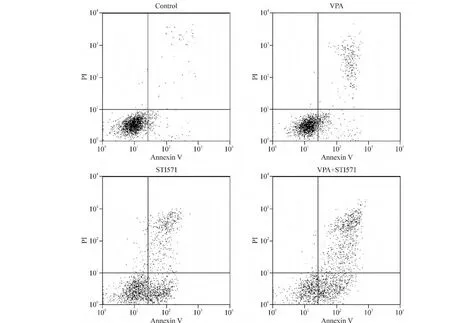
Figure 2.Effects of VPA and STI571 on K562 cell apoptosis.图2 VPA与STI571对K562细胞凋亡的影响

表2 VPA与STI571联用对K562细胞细胞周期、凋亡及Bcr/Abl mRNA表达的影响Table 2.Effects of VPA and STI571 on cell cycle,apoptosis and Bcr/Abl mRNA expression in K562 cells(Mean±SD.n=3)
讨 论
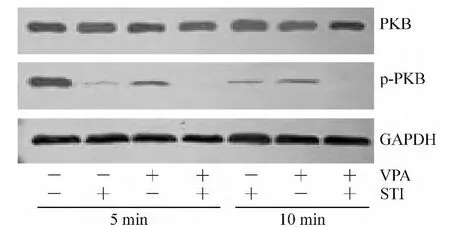
Figure 3.Impacts of VPA and STI571 on PKB and p-PKB expression in K562 cells.图3 VPA联合STI571对K562细胞PKB以及p-PKB表达的影响
表观遗传学在肿瘤发生中的作用是近年来的研究热点。科学家发现DNA启动子区过甲基化和染色质中的组蛋白去乙酰化修饰是主要的表观遗传学改变形式。因此,去甲基化药物以及HDACI也成为治疗恶性肿瘤的新型药物。去甲基化药物的代表——地西他滨,在临床治疗骨髓增生异常综合征以及难治性白血病中获得较肯定的疗效,这充分展示了靶向表观遗传学异常药物的应用前景。VPA是一种传统的抗癫痫药物,自1985年VPA被首次报道可抑制肿瘤细胞增殖后,大量的基础和临床工作者开始致力于这种新型的HADCI在抗肿瘤方面的研究[4]。我们的既往研究发现VPA能导致慢性粒细胞白血病细胞K562细胞周期阻滞并引起细胞凋亡[3,5-7]。本实验是在前期工作的基础上进一步研究VPA联合STI571对K562细胞增殖的影响及其可能机制。

表3 VPA联合STI571对K562细胞总PKB和p-PKB表达的影响Table 3.Effects of VPA and STI571 on the expression of PKB and p-PKB in K562 cells(Mean±SD.n=3)
我们的实验结果显示,VPA联合STI571对促进K562细胞的凋亡具有协同作用,这与Buchi等[8]以及Morotti等[9]研究组的报道一致,他们还发现这种协同作用在其它CML的细胞株以及CML患者的原代细胞中同样存在。我们同时检测了VPA联合STI571对K562细胞周期的影响,与对照组相比,两药合用能使细胞周期发生阻滞,主要表现为G0/G1期细胞增多,S期细胞减少。然而两药联合与单药作用相比,其阻滞作用无明显增加,说明 VPA联合STI571对细胞周期的影响无协同作用,这表明VPA增强STI571促K562细胞凋亡的作用并不是通过增加细胞周期阻滞介导的。
我们进一步检测了两药联用对Bcr/Abl融合基因的影响。实验结果表明,VPA和STI571联合处理K562细胞24 h后,VPA组以及联合用药组检测不到Bcr/Abl mRNA,而单用STI571组仍然可以检测到,而且比未加药组高,这其中的原因并不清楚,推测可能是一种药物选择下的代偿反应,这种代偿反应也有可能是临床上部分病人产生耐药的原因[10]。Buchi等[8]的结果和我们的有所不同,他们的结果显示,STI571单药处理LAMA84S细胞48h后,Bcr/Abl mRNA拷贝数轻微下降。这可能与我们处理时间为24 h,他们的实验时间为48 h以及所用的细胞株不同有关。然而,他们发现两药联用后Bcr/Abl mRNA几乎降低到零水平,这与我们的结果相似。
STI571抗CML作用的机制主要是竞争性结合Abl酪氨酸激酶催化部位的ATP结合位点,使该激酶不能与ATP结合,导致Bcr/Abl融合蛋白失去催化活性,进而阻断了异常信号转导过程[11]。现已明确,磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)信号通路活化是Bcr/Abl融合蛋白的下游信号事件之一[12]。PI3K被包括细胞因子受体或Bcr/Abl融合蛋白等酪氨酸激酶激活后,促使第二信使3,4,5-三磷酸磷脂酰肌醇(phosphatidylinositol 3,4,5-triphosphate,PIP3)形成,后者募集PKB到胞膜内侧并被其它激酶催化发生磷酸化而激活。p-PKB通过激活与细胞生长和增殖等有关的多条细胞信号通路,促进细胞的生长增殖。所以,STI571也有可能通过抑制Bcr/Abl融合蛋白的催化活性,阻断PI3K信号通路活化,从而抑制CML细胞的增殖。我们的实验结果证实了这一点,STI571处理K562细胞后,p-PKB水平明显低于对照组,而总PKB水平基本无变化。
VPA作为一种具有抗肿瘤活性的药物,其主要作用机制是抑制HDAC活性,除此之外,它还可以干扰细胞第二信使PIP3的产生和下游信号的激活[13]。基于上述理论基础,我们的实验进一步检测了VPA处理后K562细胞内p-PKB的蛋白水平。结果显示VPA可以在孵育5 min后快速下调p-PKB的蛋白水平,到孵育24 h后基本检测不到。这一结果表明VPA可能通过抑制K562细胞中PI3K信号通路的活化,减少p-PKB水平,从而干扰细胞增殖信号的转导。尽管本实验未进一步证实VPA的这种作用是直接抑制了PI3K信号,还是通过阻断Bcr/Abl信号通路而间接发挥作用的,但是我们的结果表明VPA联合STI571处理K562细胞后,其p-PKB水平较单药组更低,这说明两药在抑制PI3K信号转导方面具有协同作用。
CML疾病进展以及对STI571耐药与表观遗传学的异常有关,特别是基因甲基化和组蛋白乙酰化,阻止这2个关键过程可能可以克服患者对STI571耐药[14-15]。因此,VPA作为一种组蛋白脱乙酰酶抑制剂,从理论上讲应该可以逆转细胞对STI571的耐药。我们实验的不足之处在于未进一步研究VPA对耐STI571 CML细胞株的作用。然而,国外已经有关于此方面研究的报道。Buchi等[8]的结果令人鼓舞,他们发现VPA能协同STI571促进耐药CML细胞株LAMA84-R的凋亡,单用STI571增加Bcr/Abl mRNA的转录,而两药联用大大降低了Bcr/Abl mRNA水平。这一实验结果为VPA应用于耐STI571的CML提供了有力的实验依据。Cervera等[16]进一步尝试了VPA治疗CML的临床试验。他们用VPA和肼苯哒嗪联合STI571治疗8例(2例急变期,5例加速期,1例慢性期),对 STI571耐药的 CML患者,2例(25%)获得完全血液学缓解,1例(12.5%)获得主要细胞遗传学缓解,1例(12.5%)获得完全细胞遗传学缓解,3例(37.5%)疾病稳定,仅1例无效。以上基础和临床的实验表明了VPA在治疗耐STI571 CML方面的良好应用前景,然而仍需要更多的实验进一步确定其临床用药的适用剂量、药物搭配以及治疗疗程等,这也是我们下一步工作的研究方向。
[1] Li XN,Shu Q,Su JM,et al.Valproic acid induces growth arrest,apoptosis,and senescence in medulloblastomas by increasing histone hyperacetylation and regulating expression of p21Cip1,CDK4,and CMYC[J].Mol Cancer Ther,2005,4(12):1912-1922.
[2] Bastian L,Hof J,Pfau M,et al.Synergistic activity of bortezomib and HDACi in preclinical models of B-cell precursor acute lymphoblastic leukemia via modulation of p53,PI3K/AKT,and NF-κB[J].Clin Cancer Res,2013,19(6):1445-1457.
[3] 张祥忠,尹爱华,许多荣,等.丙戊酸钠对慢性粒细胞白血病细胞株K562增殖和凋亡的影响[J].中山大学学报:医学科学版,2008,29(5):562-565.
[4] Kostrouchová M,Kostrouch Z,Kostrouchová M,Valproic acid,a molecular lead to multiple regulatory pathways[J].Folia Biol(Praha),2007,53(2):37-49.
[5] 张祥忠,尹爱华,刘建华,等.丙戊酸钠诱导K562细胞周期阻滞和凋亡并上调 p21WAF1基因mRNA的表达[J].中国病理生理杂志,2008,24(9):1726-1729.
[6] Zhang XZ,Yin AH,Zhu XY,et al.Using an exon microarray to identify a global profile of gene expression and alternative splicingin K562 cells exposed to sodium valproate[J].Oncol Rep,2012,27(4):1258-1265.
[7] Zhang XZ,Yin AH,Lin DJ,et al.Analyzing gene expression profile in K562 cells exposed to sodium valproate using microarray combined with the connectivity map database[J].J Biomed Biotechnol,2012,2012:654291.
[8] Buchi F,Pastorelli R,Ferrari G,et al.Acetylome and phosphoproteome modifications in imatinib resistant chronic myeloid leukaemia cells treated with valproic acid[J].Leuk Res,2011,35(7):921-931.
[9] Morotti A,Cilloni D,Messa F,et al.Valproate enhances imatinib-induced growth arrest and apoptosis in chronic myeloid leukemia cells[J].Cancer,2006,106(5):1188-1196.
[10] Yu C,Rahmani M,Almenara J,et al.Histone deacetylase inhibitorspromote STI571-mediated apoptosisin STI571-sensitive and-resistant Bcr/Abl+human myeloid leukemia cells[J].Cancer Res,2003,63(9):2118-2126.
[11] Buchdunger E,Zimmermann J,Mett H,et al.Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative[J].CancerRes,1996,56(1):100-104.
[12] Skorski T,Bellacosa A,Nieborowska-Skorska M,et al.Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway[J].EMBO J,1997,16(20):6151-6161.
[13] Xu X,Muller-Taubenberger A,Adley KE,et al.Attenuation of phospholipid signaling provides a novel mechanism for the action of valproic acid[J].Eukaryotic Cell,2007,6(6):899-906.
[14] Jelinek J,Gharibyan V,Estecio MR,et al.Aberrant DNA methylation is associated with disease progression,resistance to imatinib and shortened survival in chronic myelogenous leukemia[J].PLoS One,2011,6(7):e22110.
[15] Lee SM,Bae JH,Kim MJ,et al.Bcr-Abl-independent imatinib-resistant K562 cells show aberrant protein acetylation and increased sensitivity to histone deacetylase inhibitors[J].J Pharmacol Exp Ther,2007,322(3):1084-1092.
[16] Cervera E,Candelaria M,López-Navarro O,et al.Epigenetic therapy with hydralazine and magnesium valproate reverses imatinib resistance in patients with chronic myeloid leukemia[J].Clin Lymphoma Myeloma Leuk,2012,12(3):207-212.
猜你喜欢
昆明医科大学学报(2021年3期)2021-07-22
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
中西医结合肝病杂志(2020年2期)2020-10-27
中成药(2018年3期)2018-05-07
中成药(2017年9期)2017-12-19
中成药(2017年7期)2017-11-22
中华老年多器官疾病杂志(2016年7期)2016-04-28
中国医药生物技术(2015年4期)2015-12-26
医学研究杂志(2015年9期)2015-07-01
