
基于SM 蛋白的CHO细胞分泌工程化改造和抗体表达的研究
2014-10-31 04:59刘毅华童梅刘金毅徐晨
中国医药生物技术 2014年4期
刘毅华,童梅,刘金毅,徐晨
基于SM 蛋白的CHO细胞分泌工程化改造和抗体表达的研究
刘毅华,童梅,刘金毅,徐晨
210009 南京,中国药科大学生命科学与技术学院(刘毅华);102600 北京三元基因工程有限公司(童梅、刘金毅、徐晨)
通过构建基于 SM 基因的分泌工程化改造的 CHO-AV-SM 细胞株,探讨在悬浮培养条件下,过表达转运蛋白 SM 基因对分泌蛋白表达的影响。
首先从人 HEK293FT 细胞中克隆出s1 和18c基因,然后构建含有 SM 基因的真核表达载体 pBSM;随后将 pBSM 质粒稳定转染入 CHO-AV 细胞并鉴定,获得了基于 SM 基因的分泌工程化改造的 CHO-AV-SM 细胞株;在此基础上对 CHO-AV-SM 细胞株进行悬浮培养适应,并测定了 CHO-AV 和 CHO-AV-SM 细胞株的生长和蛋白表达参数。
过表达转运蛋白 SM 基因,CHO-AV-SM 细胞株的抗体表达量较 CHO-AV 抗体表达量提高约 75%,且测得第 6 天细胞数达到最大值,此时 CHO-AV 的表达量约为 45.4 μg/ml,CHO-AV-SM 的表达量为 79.5 μg/ml。
通过构建全抗稳定表达细胞,在细胞中过表达 SM 基因,并悬浮培养适应,建立了悬浮培养条件下基于转运蛋白过表达的分泌工程改造方法,为转运蛋白改造 CHO 细胞,提升全抗表达能力提供了有力支持。
CHO 细胞; 蛋白质工程; SLY1 蛋白质类; MUNC18c蛋白质类; 贝伐单抗
分泌表达是真核细胞表达基因工程重组蛋白的主要表达途径,在外源基因拷贝数及其转录水平达到瓶颈时,提高细胞分泌能力有助于重组蛋白表达量的提升。Sly1/Munc18(SM)蛋白是一类高度保守的囊泡转运蛋白家族,其中Sly1 调控囊泡由高尔基体至细胞膜的转运,而Munc18c则调控囊泡由内质网至高尔基体的转运[1]。已有研究表明在酵母和人细胞中过表达 SM 蛋白能够提高分泌蛋白的表达量[2]。研究人员把这种基于转运蛋白过表达的细胞改造称为“分泌工程化改造”[3]。
本研究从人 HEK293FT 细胞中克隆s1 和18c基因,并构建含有 SM 基因的真核表达载体;然后将构建好的 SM 基因共表达质粒 pBSM 导入以贝伐单抗为报告基因的 CHO-AV 细胞株中,借以构建稳定过表达SM 基因的细胞株CHO-AV-SM。通过运用高通量筛选法获得外源蛋白表达量较高的工程细胞株,并对工程细胞株进行悬浮培养适应。在此基础上,通过比较 CHO-AV- SM 细胞与 CHO-AV 细胞在悬浮培养条件下的生长代谢差异,进而验证 SM 蛋白促进 CHO 细胞分泌表达外源蛋白的功能,并为构建分泌工程化改造细胞确立可行的研究方向。
1 材料与方法
1.1 材料
1.1.1 细胞系 CHO-K1 及 HEK293FT 细胞购自 ATCC;CHO-AV 细胞由北京三元基因工程有限公司构建。
1.1.2 试剂 限制性内切酶、T4 DNA 连接酶及 PCR 聚合酶购自宝生物工程有限公司;质粒大提、总 DNA、RNA 提取及 TIANScript cDNA 第一链合成试剂盒购自天根生化科技有限公司;Lipofactamine2000、pBudCE4.1、pcDNA3.1(+) 及 pVITRO2-neo-mcs 质粒、DMEM/F12、CHO 化学限定培养基及胎牛血清购自美国 Invitrogen 公司;pcDNA3.1-AVL(轻链)、pcDNA3.1-AVH(重链)及 pBSM 质粒由北京三元基因工程有限公司构建;HRP 标记羊抗人 κ 二抗购自美国Southern Biotech 公司;羊抗人 Fc 多抗购自美国KPL 公司。
1.1.3 仪器 PTC-100TM 型 PCR 扩增仪为美国 MJ Research 公司产品;细胞恒温振荡培养箱为瑞士Adolf Kuhner AG 公司产品;DSZ5000X 型倒置生物显微镜购自重庆澳浦光电技术有限公司。
1.2 方法
1.2.1 SM 基因的克隆 收集培养 72 h 的HEK293FT 细胞,细胞计数,用 PBS 洗涤 3 次,参照总 RNA 提取试剂盒说明提取总 RNA,样品溶解于 50 μl RNase-free ddH2O 中,1% 琼脂糖凝胶电泳检测总 RNA 纯度,紫外分光光度测定浓度。参照 TIANScript cDNA 第一链合成试剂盒说明书,以获得的总 RNA 为模板,以 Oligo(dT) 为引物反转录合成 cDNA,以 cDNA 作为模板,分别以1 基因的引物1、2 及18c基因的引物18c1、18c2进行 PCR 扩增,扩增产物进行 1.5% 琼脂糖凝胶电泳分析,并用 1.5% 琼脂糖凝胶电泳进行酶切产物分离。切下 1500~2000 bp 之间区域的目的 DNA 片段1 基因和18c基因,并用 DNA 片段回收试剂盒纯化线性目的片段。1 及18c基因的上下游引物序列如下:1 上游:5' CCGCTCGAGAC CATGGCGGCGGCGGCGGCAGCG 3',下游:5' CG CGGATCCTTACTTTTGTCCAAGTTGTGACAACTG 3';18c1 上游:5' CCGCTCGAGACCATGGC GCCGCCGGTGGCAGAGAGG 3',下游:5' CGCGG ATCCCTATTCATCTTTAATTAAGGAGAC 3'。
1.2.2 SM 基因共表达载体的构建 分别将上述回收的1 基因和18c基因以I 和H I 进行双酶切,酶切完成后,用 1.5% 琼脂糖凝胶电泳进行酶切产物分离,切下 1500 ~2000 bp之间区域的目的 DNA 片段,并用 DNA 片段回收试剂盒纯化线性目的片段。使用 T4 DNA 连接酶连接1 基因片段和以II、I 双酶切的真核表达载体 pBudCE4.1,将获得的重组质粒转化 Top10 感受态细胞,挑选测序正确的克隆,并进行质粒提取,得到重组质粒 pBudCE4.1-1命名为 pBS。将真核表达载体 pBudCE4.1 和 pBudCE4.1-1 以H I、I 进行双酶切,使用 T4 DNA 连接酶连接双酶切后的18c基因片段和真核表达载体 pBudCE4.1 和 pBudCE4.1-1,将获得的重组质粒转化 Top10 感受态细胞,挑选测序正确的克隆,并进行质粒提取,将重组质粒 pBudCE4.1-18c 和 pBudCE4.1-1-18c分别命名为 pBM 和 pBSM。
1.2.3 SM 基因共表达载体转染 CHO-AV 细胞株 采用 Lipofactamine2000 转染试剂进行稳定转染。将 2 × 106个 CHO-AV 细胞接种 T-25 方瓶,当 CHO-AV 细胞密度在 80% ~ 90%时进行转染。将培养基换成 5 ml 无血清 DMEM/F12 培养基,制备质粒-培养基混合物(质粒 10 μg +无血清培养基 500 μl),同时配置转染试剂-培养基混合物(转染试剂 25 μl +无血清培养基 500 μl),上述混合物分别静置 5 min 后进行混合,静置 20 min 后加入上述细胞内,37 ℃,5% CO2培养 6 h,将培养基换成 6 ml 5% 胎牛血清 DMEM/F12 培养基继续培养,并运用 Zeocin 和有限稀释法进行单克隆筛选。
1.2.4 外源 SM 蛋白表达的鉴定 收集培养 72 h的转染 SM 基因的 CHO-AV 细胞,细胞计数,用 PBS 洗涤 3 次,参照动物细胞总 DNA、RNA、蛋白分离试剂盒的说明分别提取基因组 DNA、总 RNA 和总蛋白,用于 SM 基因的 DNA 水平、mRNA 水平及蛋白水平的检测。在进行基因组 DNA 水平检测时,以基因组 DNA 作为模板,以引物12以及引物18c118c2 进行 PCR 扩增,产物进行 1.5% 琼脂糖凝胶电泳分析。在进行 mRNA 水平检测时,参照 TIANScript cDNA 第一链合成试剂盒,以上步骤中获得总 RNA 为模板,以 Oligo(dT) 为引物逆转录合成 cDNA;以 cDNA 作为模板,以引物1、2及引物18c1、18c2 进行 PCR 扩增,扩增产物进行 1.5% 琼脂糖凝胶电泳分析。在进行蛋白水平检测时,以 CHO-K1 细胞总蛋白为阴性对照,并将提取的总蛋白进行 SDS-PAGE 电泳及 Western blot 鉴定。一抗为鼠抗人sly1 抗体和羊抗人munc18c抗体,二抗为 HRP 兔抗鼠多抗和 HRP 驴抗羊多抗。
1.2.5 ELISA 筛选高表达细胞株 运用双抗体夹心 ELISA 测定上清抗体浓度,包被抗体为羊抗人 IgG Fc 抗体,标准品为人 IgG,酶标二抗为羊抗人 κ 链抗体。从中挑选出抗体表达量最高的一株细胞命名为 CHO-AV-SM,进行悬浮适应。
1.2.6 悬浮培养 CHO-AV-SM 细胞与 CHO-AV 细胞表达、生长曲线的测定和比较 CHO-AV-SM 以及 CHO-AV 以 3 × 105个/ml初始细胞密度接种于 30 ml 无血清培养基,37 ℃、5% CO2、110 r/min 培养。每 24 小时取一次样(约 500 μl),台盼蓝染色计活细胞数及总细胞数,同时离心冻存上清。当细胞活力低于 50% 时,终止细胞培养。ELISA 检测各时间点样品抗体浓度。计算抗体比产率,绘制活细胞数-时间、细胞存活率-时间、抗体浓度-时间、抗体比产率-时间曲线图。
1.3 统计学处理
2 结果
2.1 SM 基因的克隆
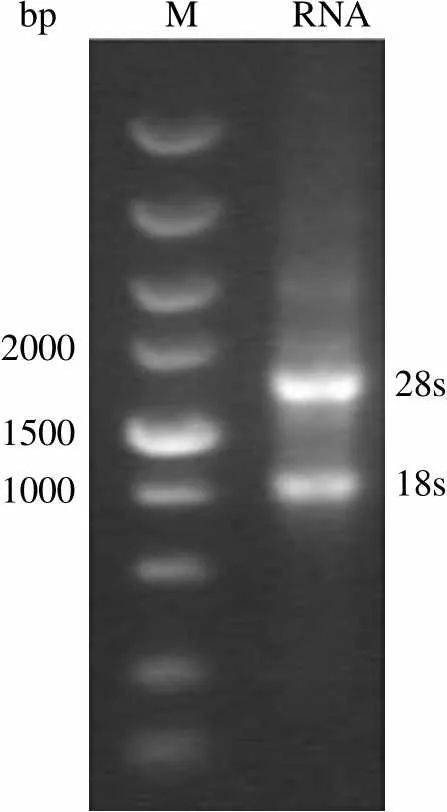
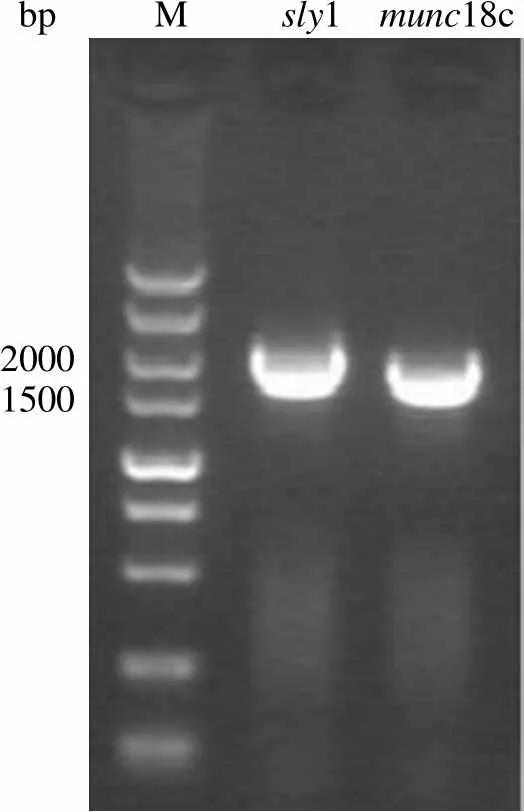
从 HEK293FT 中提取总 RNA 的电泳结果如图 1 所示。图中 RNA 可见 28s 和 18s 两条带且 28s 带亮度超过 18s,说明所提 RNA 基本未降解;紫外分光光度法检测总 RNA 浓度为 601 ng/ml。使用1 基因引物和18c基因引物分别进行 RT-PCR 后,电泳结果如图 2 所示,1 片段电泳条带在约 1930 bp,18c片段电泳条带约在 1780 bp,大小符合预期。
bp M RNA 2000 15001000 28s 18s
Figure 1 Total RNA of HEK293FT cells
2.2 共表达载体鉴定
pBudCE4.1-1、pBudCE4.1-18c 和 pBudCE4.1-1-18c 质粒示意图如图 3所示。对构建的共表达载体 pBudCE4.1-1、pBudCE4.1-18c和pBudCE4.1-1-18c 进行 PCR 鉴定,结果如图 4 所示。1 片段和18c片段的电泳条带分别约 1930 bp 和 1780 bp,大小符合预期。pBS 大提质粒浓度为504 ng/μl,回收体积 1.5 ml;pBM 大提质粒浓度为 560 ng/μl,回收体积 1.5 ml;pBSM 大提质粒浓度为 613 ng/μl,回收体积 1.5 ml。
bp M sly1 munc18c 20001500
Figure 2 To clone1 and18c gene by RT-PCR from total RNA of HEK293 cells
2.3 SM 基因共表达载体转染 CHO-AV 细胞株
2.3.1 Zeocin 筛选细胞状态观察及有限稀释法挑单克隆 24 h 后按照 1:10 消化传代,48 h 后加 Zeocin 培养。每 3 天换一次培养基,维持 Zeocin 浓度在 200 μg/ml,加压筛选 12 d。从加 Zeocin 开始 4 ~ 5 d 内,细胞状态正常;5 d 后细胞开始大量死亡,具体表现为细胞变大膨胀,有些细胞形状由梭形变成不规则形,部分细胞有明显空泡形成,有死细胞漂浮在培养基中(图 5A)。10 d后具有抗性的单个细胞克隆形成(图 5B)。至 12 d 将所有细胞克隆消化,有限稀释法挑单克隆。铺板 6 ~ 7 d 后在镜下可看到明显的单克隆形成(图 5C)。
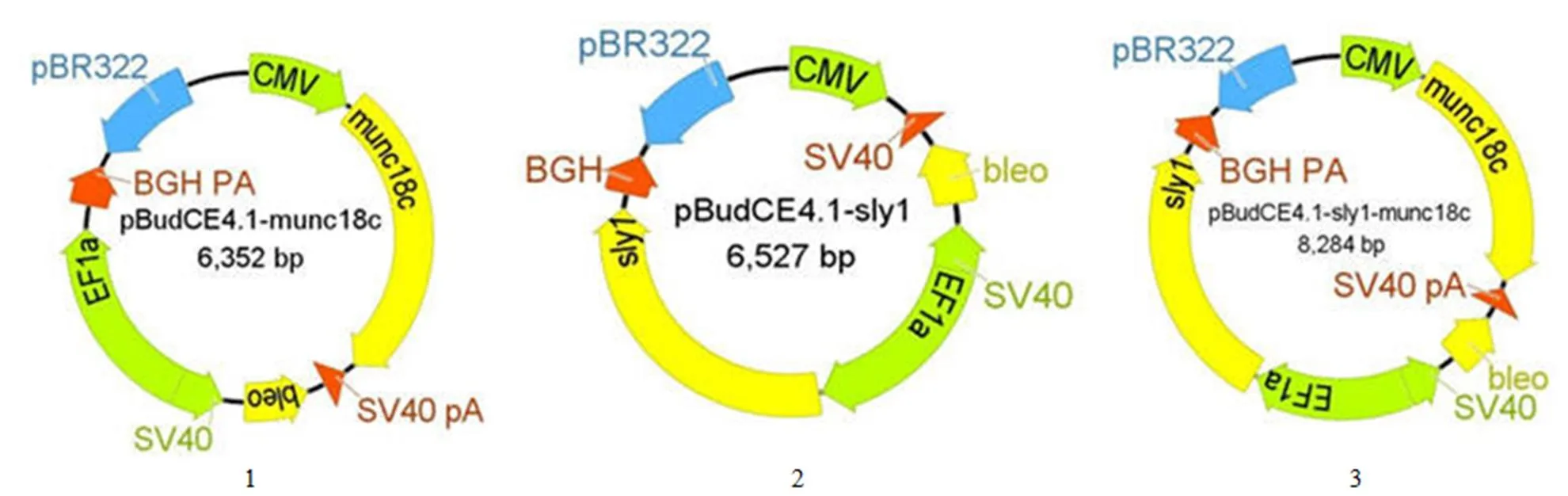
图 3 质粒结构示意图(1:pBudCE4.1-sly1;2:pBudCE4.1-munc18c;3:pBudCE4.1-sly1-munc18c)
Figure 3 Schematic diagrams of plasmid (1: pBudCE4.1-1; 2: pBudCE4.1-18c; 3: pBudCE4.1-1-18c)
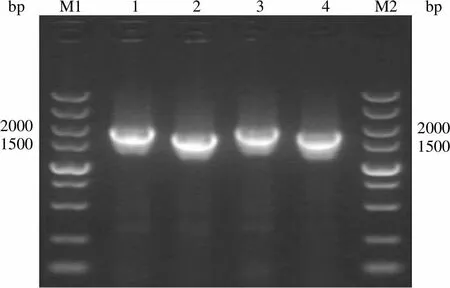
bp M1 1 2 3 4 M2bp 20001500 20001500
Figure 4 Determination of1 and18c PCR fragments of plasmid pBudCE4.1-1, pBudCE4.1-18c and pBudCE4.1-1-18c
2.3.2 SM 基因表达的鉴定
2.3.2.1 基因组水平及转录水平鉴定 收集培养 72 h 的细胞抽提基因组 DNA、总 RNA 和总蛋白,将总 RNA 反转录为 cDNA,以 gDNA 和 cDNA 为模板,分别用1 基因引物1、2 以及18c基因引物18c1、18c2 进行 PCR 扩增,结果如图 6 ~ 7。1 片段电泳条带约在 1930 bp,18c片段电泳条带约在 1780 bp,大小符合预期。
2.3.2.2 蛋白水平鉴定 CHO-AV 总蛋白为阴性对照,上样量均为 20 μl,进行 SDS-PAGE 电泳,使用抗人sly1 抗体和抗人 munc18c 抗体进行Western blot 检测,结果如图 8 所示。sly1蛋白理论分子量为72.3 kD,munc18c理论分子量为 67.7 kD。结果显示 5 株稳定转染细胞均有人源sly1 和 munc18c 蛋白的表达,CHO-AV 细胞自身表达的sly1 和 munc18c是非人源蛋白,理论上检测不到,在sly1 和munc18c蛋白分子量理论值附近没有条带。
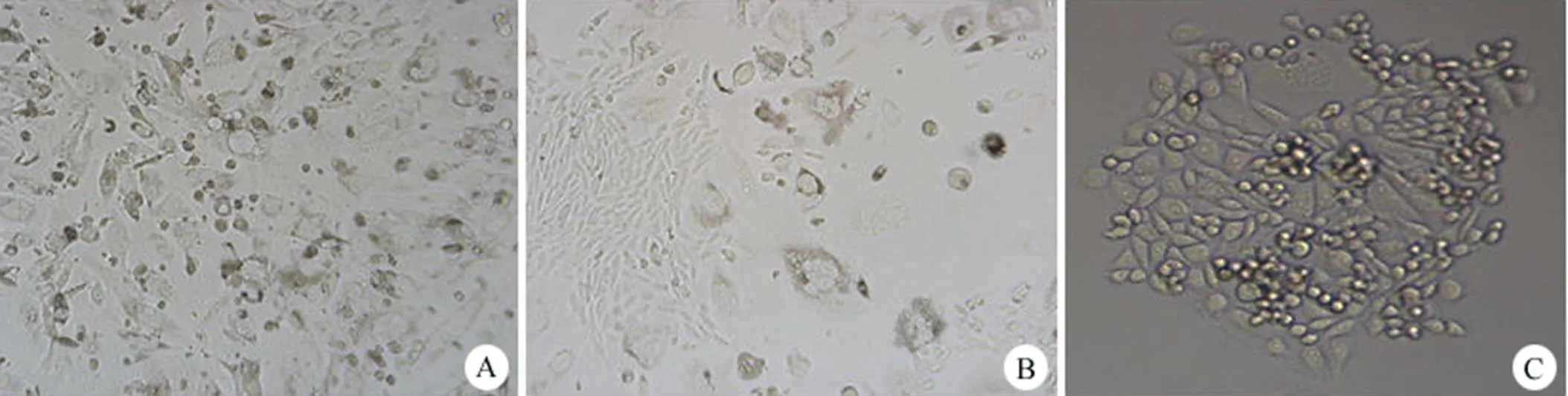
图 5 镜下观察 Zeocin 筛选细胞状态及单克隆细胞形成(A:细胞因 Zeocin 筛选大量死亡;B:抗性细胞克隆形成;C:单克隆示意图)
Figure 5 Cellular morphology and single clones during Zeocin screening under microscope detection (A: Large number of cells died during Zeocin screening; B: Zeocin resistant cell colony appeared; C: Microscope detection of single colony)
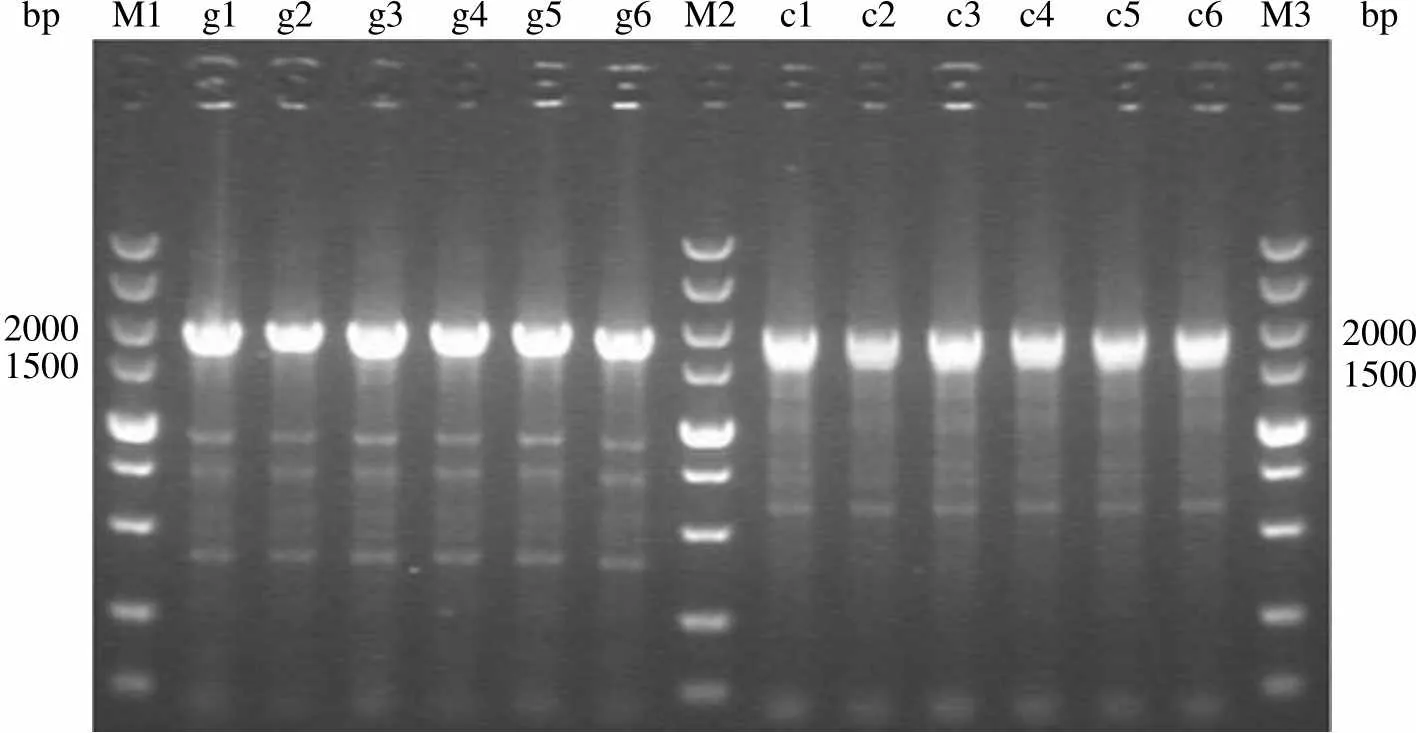
bp M1 g1 g2 g3 g4 g5 g6 M2 c1 c2 c3 c4 c5 c6 M3bp 20001500 20001500
Figure 6 Fragments of1 from gDNA and cDNA of stable expression cells
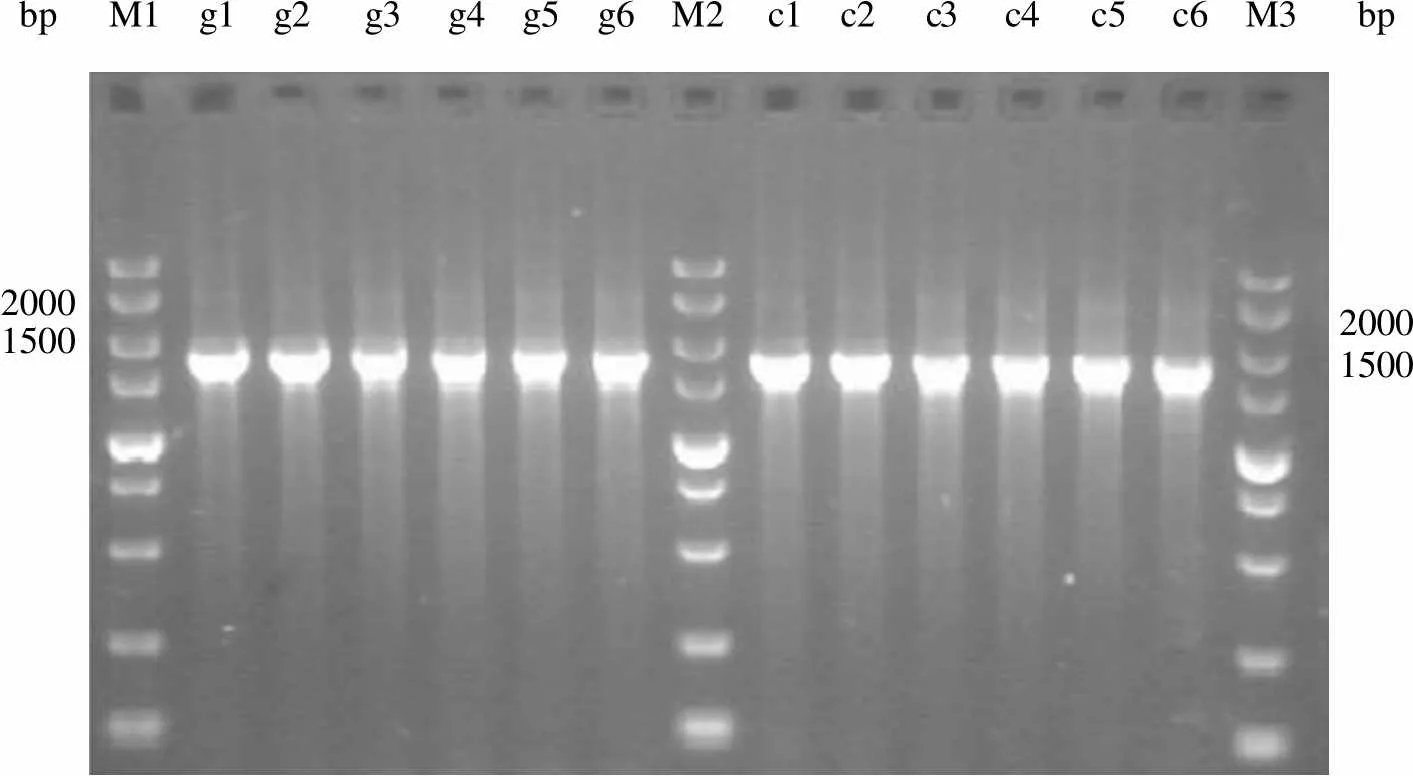
bp M1 g1 g2 g3 g4 g5 g6 M2 c1 c2 c3 c4 c5 c6 M3bp 20001500 20001500
Figure 7 Fragments of18c from gDNA and cDNA of stable expression cells
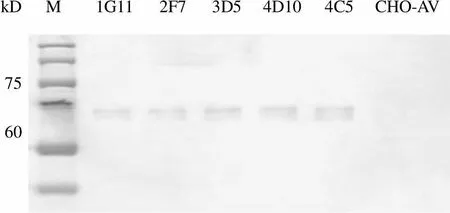
kD M 1G11 2F7 3D5 4D10 4C5 CHO-AV 75 60
Figure 8 Western blot of sly1 and munc18c from different clones
2.4 ELISA 筛选高表达单克隆细胞株
从有限稀释的 4 块 96 孔板中挑选形态较好的 5 株单克隆扩大培养,编号为(96 孔板编号 + 各孔编号)1G11、2F7、3D5、4D10、4C5 并冻存。6 孔板到 T-25 瓶扩大过程中收取上清,以 CHO-AV 表达上清为阴性对照进行检测。结果如表 1。将表达量最高的细胞株 CHO-4C5命名为 CHO-AV-SM,接入摇瓶悬浮适应培养。
2.5 悬浮培养 CHO-AV-SM 细胞与 CHO-AV 细胞表达、生长曲线的测定和比较
CHO-AV-SM 和 CHO-AV 以 3 × 105个/ml 初始细胞密度接种于 30 ml CD-CHO 培养基,每 24 小时取样,以台盼蓝染色计活细胞数及总细胞数,同时离心冻存上清。12 d 后终止细胞培养。ELISA 检测各时间点样品抗体浓度。计算抗体比产率,公式如下:
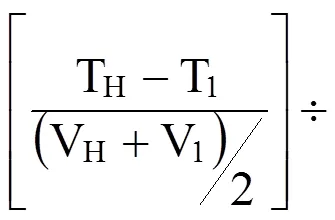
公式中 qMab:抗体比产率;TH:抗体最终浓度(μg);TI:抗体初始浓度(μg);VH:最终细胞数(106cells);VI:接种细胞数(106cells);t:时间(d)。
绘制活细胞数-时间、细胞存活率-时间、抗体浓度-时间、抗体比产率-时间曲线图。如图 9 所示,CHO-AV 细胞在批培养条件下前 6 天细胞活力维持在约 100%,且第 6 天细胞数达到最大值,之后下降,到第 12 天细胞活力已低于 50%;抗体浓度一直在增加,但抗体比产率呈现逐渐下降趋势。与参考文献结果相比,CHO-AV 生长速率及最大密度有较大差距。
所谓“阳光是最好的防腐剂”⑩,透明度在规则制定中一直占有重要地位。透明度在公开和保密之间形成张力:政府机构或企业为追求透明度而公开信息,可能会损害国家安全或商业秘密等重要利益,不充分的透明度则会导致信任缺失,而无意的披露又可能造成损害隐私或泄露机密等不利后果。有研究提出大数据的“透明悖论”⑪,即某些机构为了执行任务或提供服务,会运用法律和商业秘密武器来隐匿其数据收集行为及收集的数据,因而如何发现数据收集行为并要求其实现透明度呢?
同 CHO-AV 细胞相比,CHO-AV-SM 细胞参数变化趋势基本一致,如图 9 所示,前 6 天细胞活力维持在约 100%,第 6 天细胞数达到最大值,之后下降,到第 12 天细胞活力已低于 50%;抗体浓度一直增加,但抗体比产率呈现逐渐下降趋势。这提示插入新的外源基因会影响细胞的生长,但验证了在宿主细胞过表达sly1和munc18c的促分泌表达作用。这两株细胞的细胞密度均在培养第 6 天达到最大值,此后细胞大量死亡。由于死细胞裂解物给培养上清带来许多杂蛋白,影响产物质量。因此我们以第 6 天抗体浓度进行比较:此时 CHO-AV 细胞的抗体表达量约为 45.4 μg/ml;CHO-AV-SM 细胞的抗体表达量为 79.5 μg/ml,在 CHO-AV 基础上提高约 75%,这与贴壁培养数据接近。

表 1 部分高表达细胞株抗体表达量
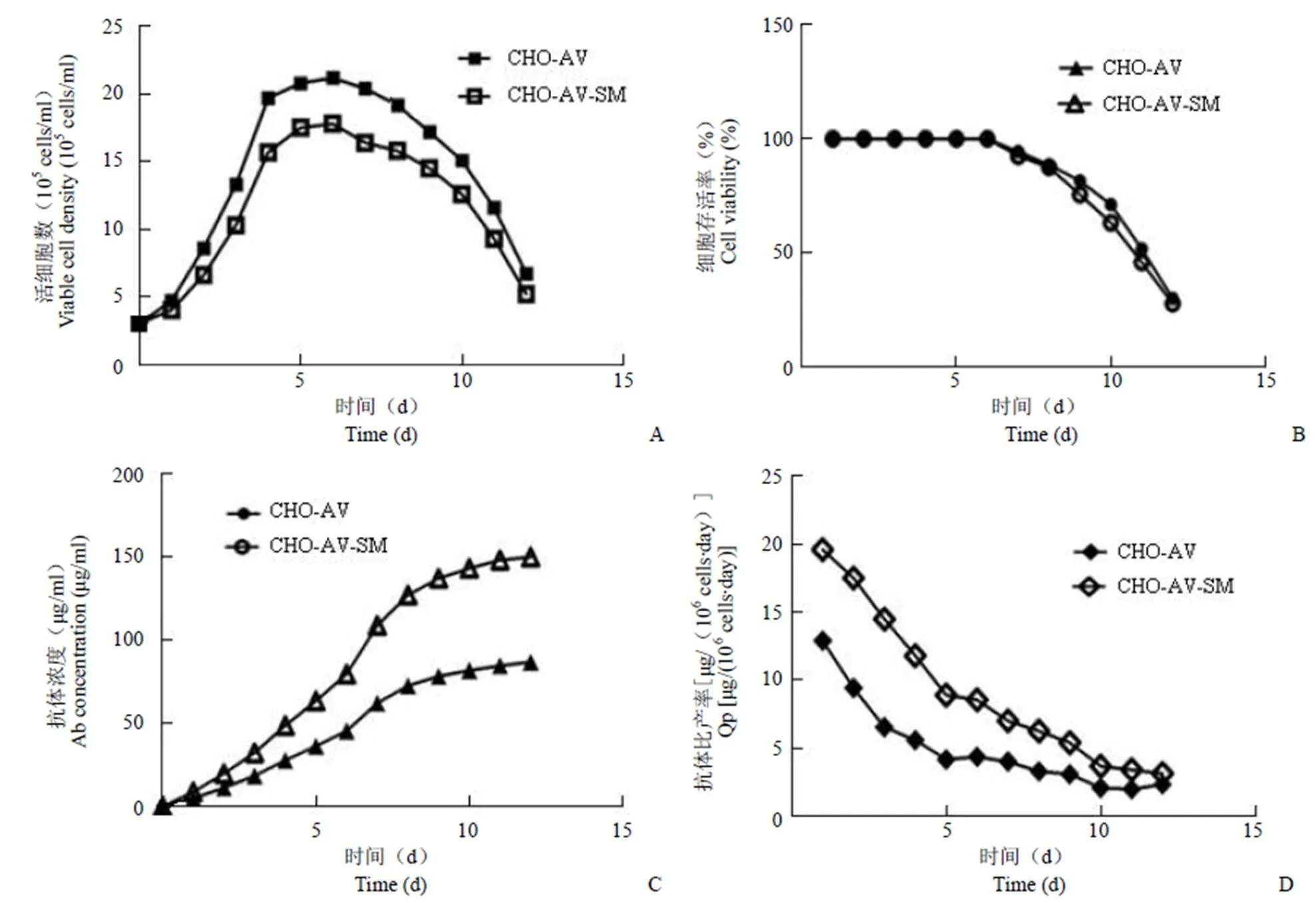
图 9 悬浮培养的 CHO-AV-SM 与 CHO-AV 细胞的活细胞数-时间(A)、细胞存活率-时间(B)、抗体浓度-时间(C)、抗体比产率-时间(D)曲线对比
Figure 9 The comparison of viable cell density (A)、cell viability (B)、antibody titer (C) and Qp (D) between CHO-AV-SM and CHO-AV
将 CHO-AV-SM 细胞数据和 CHO-AV 细胞数据对比,可以看出 CHO-AV 细胞的生长速率及最大密度略高于 CHO-AV-SM 细胞,但是 CHO-AV 细胞的抗体产量和抗体比产率低于 CHO-AV-SM 细胞。
3 讨论
分泌表达是真核细胞表达基因工程重组蛋白的主要表达途径。经研究表明有多种因素影响抗体基因在 CHO 细胞中稳定表达,包括抗体基因在宿主染色体上的整合位点、基因拷贝数、抗体mRNA的转录效率和 mRNA 的稳定性、抗体基因的翻译、抗体轻重链的组装、分泌以及抗体本身结构等多个方面。在外源基因拷贝数及其转录水平达到蛋白转运能力的上限时,提高细胞分泌能力有助于重组蛋白表达量的提升。囊泡的转运和分泌实质上是胞内膜分离和融合的循环过程。研究发现,所有囊泡转运途径中的胞内膜融合过程都需要 SNARE 蛋白家族和SM蛋白家族协同参与调节。其中 SM 蛋白是一类亲水性蛋白质,由 600 个左右氨基酸组成,其空间结构大致为“弓形”结构蛋白[4]。利用 SM 蛋白促进细胞分泌的特性,近年来人们开始尝试以介导胞内大分子运输的转运蛋白为靶点提高重组蛋白表达量并取得了突破。Peng和Fussenegger[5]的研究表明,在 CHO 细胞中过表达sly1可以使利妥昔单抗的表达量提高 10 倍,同时表达Sly1和Munc18c则可以使其产量提高 15 倍。Hou 等[6]构建了稳定表达 SLY1 和 SEC1 的酿酒酵母并分别用来表达异源蛋白(人胰岛素前体、曲霉α-淀粉酶)和内源蛋白(转化酵素),结果各个目的蛋白表达量均有不同程度的提升。由于 CHO 细胞与人细胞的 SM 基因具有较高同源性,推测其 SM 基因调控囊泡转运机制与人细胞类似,所以本课题选择从人细胞中克隆 SM 基因并进行后续研究。
本研究将含有1 和18c基因的真核共表达载体导入 CHO-AV 细胞株,在研究中首先从人细胞克隆了1 基因和18c基因并构建表达载体 pBS、pBM 和 pBSM。通过 SM 基因的稳定转染,我们获得的基于 SM 基因的分泌工程化改造的 CHO 细胞株 CHO-AV-SM 与原细胞株 CHO-AV 抗体表达量相比提高约 75%,但与参考文献中 SM 促表达效果相比还有差距。这可能是因为筛选得到的 CHO-AV 细胞表达量处于较低水平,转运途径的瓶颈效应对其影响较小,因此,SM 基因无法表现出较好的促表达效果。在后续工作中应该对载体元件和筛选方法进行系统优化,与分泌途径优化一起协同提高 CHO 工程细胞株的抗体表达量[7]。从图 9可以看出,CHO-AV 细胞及 CHO-AV-SM 细胞在批悬浮批培养条件下前 6 天细胞活力维持在近 100%,且第 6 天细胞数达到最大值,之后下降,到第 13 天细胞活力已低于 50%;抗体浓度持续增加,但比产率呈现逐渐下降趋势。与参考文献结果相比,各个参数的变化趋势大致相同,但生长速率及最大密度低于参考文献结果。CHO-AV 细胞和 CHO-AV-SM 细胞相比,CHO-AV 细胞的生长速率及最大密度略高于 CHO-AV-SM细胞。一种可能是因为稳定转染后,外源基因随机插入基因组,有可能破坏宿主细胞正常功能的序列,造成不同细胞单克隆之间生长特性的差异;另一种可能则是外源蛋白的表达增加了宿主细胞的代谢负担,更多的营养物质被用来表达而非生长。所以在筛选过程中不但要考察细胞的表达量,还应该考察细胞的生长情况,选择表达量高且生长情况良好的细胞株进行后续开发[8]。另外也可采用定点整合技术,将外源基因定向插入 CHO 细胞基因组的高表达位点,从而提高筛选成功率[9]。
[1] Ueberberg S, Schneider S. Phage library-screening: a powerful approach for generation of targeting-agents specific for normal pancreatic islet-cells and islet-cell carcinoma in vivo. Regul Pept, 2010, 160(1-3):1-8.
[2] Peng RW, Guetg C, Tigges M, et al. The vesicle-trafficking proteinincreases the secretory capacity of mammalian cells. Metab Eng, 2010, 12(1):18-25.
[3] Sommavilla R, Lovato V, Villa A, et al. Design and construction of a naïve mouse antibody phage display library. J Immunol Methods, 2010, 353(1-2):31-43.
[4] Ki MK, Kang KJ, Shim H. Phage display selection of EGFR-specific antibodies by capture-sandwich panning. Biotechnol Bioprocess Eng, 2010, 15(1):152-156.
[6] Hou J, Tyo K, Liu Z, et al. Engineering of vesicle trafficking improves heterologous protein secretion in Saccharomyces cerevisiae. Metab Eng, 2012, 14(2):120-127.
[7] Rita Costa A, Elisa Rodrigues M, Henriques M, et al. Guidelines to cell engineering for monoclonal antibody production. Eur J Pharm Biopharm, 2010, 74(2):127-138.
[8] Rodrigues ME, Costa AR, Henriques M, et al. Technological progresses in monoclonal antibody production systems. Biotechnol Prog, 2010, 26(2):332-351.
[9] Hansel TT, Kropshofer H, Singer T, et al. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov, 2010, 9(4):325-338.
High expression of antibody in secretion engineered CHO cells based on SM protein
LIU Yi-hua, TONG Mei, LIU Jin-yi, XU Chen
The aim of this study is to investigate the improvement of secreted protein expression by constructing antibody-producing CHO cells based on molecular engineering of secretion, which includes SM gene over-expression and cell adaptation to suspension culture.
In this study, we first cloned1 gene and18c gene from human cells and constructed the eukaryotic co-expression vector. We then successfully transfected the1 and18c genes into CHO-AV cells, and identified the SM protein expressing CHO-AV-SM cells. We next suspended the adapted CHO-AV-SM cells in culture and determined the growth and the expression parameters of CHO-AV and CHO-AV-SM cells.
Due to SM transporter protein gene over-expression, antibody expression of CHO-AV-SM cell line was increased by 75%,as compared with CHO-AV cells. On the sixth day the cell number reached the maximum, and the antibody production of CHO-AV and CHO-AV-SM cells was about 45.4 μg/ml and 79.5 μg/ml, respectively.
This study improves the antibody production of CHO cells by molecular engineering of secretion, which includes SM gene over-expression and cell adaptation to suspension culture. This provides supports to molecular engineering of CHO-cell secretion and antibody production improvement.
CHO cells; Protein engineering; SLY1 proteins; MUNC18C proteins; Bevacizumab
XU Chen, Email: xuchen@triprime.com
10.3969/cmba.j.issn.1673-713X.2014.04.003
徐晨,Email:xuchen@triprime.com
2014-05-16
AuthorAffiliations: College of Life Science and Technology, China Pharmaceutical University, Nanjing 210009, China (LIU Yi-Hua); Beijing Tri-Prime Genetic Engineering Co., Ltd, Beijing 100260, China (TONG Mei, LIU Jin-yi, XU Chen)
猜你喜欢
成都医学院学报(2022年4期)2022-08-19
城市道桥与防洪(2022年3期)2022-05-08
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
江西农业学报(2021年4期)2021-04-20
安全与环境工程(2021年2期)2021-04-02
中西医结合肝病杂志(2020年2期)2020-10-27
煤炭加工与综合利用(2020年6期)2020-07-17
三农资讯半月报(2020年11期)2020-06-21
医学研究杂志(2015年9期)2015-07-01
