
间甲基苯甲醚分子旋转异构体的质量分辨阈值电离光谱
2014-10-18 05:28曾圣渊曾文碧
物理化学学报 2014年8期
秦 晨 曾圣渊 张 冰 曾文碧,*
(1台湾中央研究院原子与分子科学研究所,生物分子质谱与光谱学实验室,台北10617;2中国科学院武汉物理与数学研究所,波谱与原子分子物理国家重点实验室,武汉 430071;3新疆师范大学物理与电子工程学院,新型发光材料与纳米微结构实验室,乌鲁木齐 830054)
1 Introduction
Scientists have found that rotational isomers(rotamers)involve in some biological phenomena and processes.1,2Oikawa et al.3reported that some meta substituted anisoles and phenols have rotamers coexisting in chemical samples.Theoretical calculations predict that m-methylanisole has two stable structures,as shown in Fig.1.Both the methyl and methoxyl groups can donate electrons to the aromatic ring through the σ bond.In addition,the CH3group can interact with the π electrons of the ring through hyperconjugation and the OCH3group can share its lone-pair electrons in the oxygen atom with the ring.4These substituent-ring interactions will alter the electron density nearby the aromatic ring,leading to some changes in the molecular geometry and vibrations in the electronic state.m-Methylanisole is an important intermediate in organic synthesis and is mainly used as intermediate in the synthesis of dyestuffs and pharmaceuticals.This molecule has been studied by various spectroscopic methods.5-9Breen et al.5performed the excitation and dispersed laser-induced fluorescence(LIF)and resonance enhanced multiphoton ionization time-of-flight(REMPI TOF)mass spectroscopy experiments.They reported the origins of the S1←S0electronic transition of two rotamers and the energy barriers to the CH3torsion of m-methylanisole in the ground S0and electronically excited S1states.Ichimura,Suzuki,and their coworkers6,7recorded the excitation and dispersed LIF and hole-burning spectra.They also performed ab initio calculations to confirm the existence of the cis and trans rotamers of m-methylanisole.In addition to the investigations of the methyl torsion,they found three vibronic bands resulting from in-plane ring deformation vibrations in the S1state.Alvarez-Valtierra et al.8applied rotationally resolved fluorescence excitation spectroscopy and reported the rotational constants in the S0and S1states and the precise S1←S0electronic transition energies of the cis and trans rotamers of m-methylanisole.Concerning the ionic properties,Fujii et al.9applied pulsed field ionization zero kinetic(PFI-ZEKE)photoelectron spectroscopy to investigate the CH3torsion of o-,m-,and p-methylanisole in the D0state.To the best of our knowledge,the experimental data related to ring vibrations of the selected rotamers of m-methylanisole cation are still not available in the literature.Because many ring vibrations are active in most of benzene derivatives,their spectroscopic information is useful for molecular identification.

Fig.1 Molecular structures of(a)cis-m-methylanisole and(b)trans-m-methylanisole
In the present study,we apply the one-color and two-color resonant two-photon ionization(1C-and 2C-R2PI)and massanalyzed threshold ionization(MATI)techniques to investigate the molecular properties of m-methylanisole in the S1and cationic ground D0states.The MATI spectra of the selected rotamers were recorded by ionizing through a few intermediate vibronic states in the S1state to obtain more information about the active cation vibrations.Comparison of the experimental data of m-methylanisole with those of anisole,10toluene,11and their derivatives12-21allows one to learn the substitution effects of the methyl and methoxyl groups on the transition energy and molecular vibration.We have also performed the ab initio and density function theory(DFT)calculations to provide possible interpretation of our experimental findings.Because the present experiments involve twophoton excitation and ionization steps,these spectroscopic data are useful to gain knowledge about photochemical and photophysical processes with UV light sources.
2 Experimental and computational methods
2.1 Experimental method
The experiments reported here were performed by using a laserbased TOF mass spectrometer,described in our previous publication.22Only a brief description is stated here.m-Methylanisole(99%purity)was purchased from Sigma-Aldrich Corporation and used without further purification.The sample was heated to about 50°C to acquire sufficient vapor pressure.The vapors were seeded into 0.2-0.3 MPa of helium and expanded into the vacuum through a pulsed valve with a 0.15 mm diameter orifice.The generated molecular beam was collimated by a skimmer located 15 mm downstream from the nozzle orifice.During the experiments,the gas expansion,laser-molecular beam interaction,and ion detection regions were maintained at pressures of about 1×10-3,1×10-5,and 1×10-6Pa,respectively.
The photoionization efficiency(PIE)and MATI experiments are accomplished by applying two independent tunable UV lasers controlled by a delay/pulse generator(Stanford Research Systems DG535).The excitation source is a Nd:YAG pumped dye laser(Quanta-Ray PRO-190-10/Lambda-Physik,Scanmate UV with BBO-I crystal;Coumarin-540 dye).The generated visible radiation is frequency-doubled to produce UV radiation.The ionization UV laser(Lambda-Physik,Scanmate UV with BBO-III crystal;LDS-698 and DCM dyes)is pumped by a frequencydoubled Nd-YAG laser(Quanta-Ray LAB-150).A laser wavelength meter(Coherent,WaveMaster)was used to calibrate the wavelength of laser.Due to the systematic instrument errors,to obtain the true wavelength,the displayed wavelength of the excitation and ionization lasers needs to be added by 0.20 and 0.34 nm,respectively.The resulting values are nearly identical with those of the high resolution electronic spectroscopic experiments.8,23These two counter-propagating laser beams were focused and intersected perpendicularly with the molecular beam at 50 mm downstream from the nozzle orifice.
The PIE experiment is done by scanning the ionization(probe)laser while fixing the excitation(pump)laser at a particular intermediate(e.g.S100)level of the selected molecular species.It detects non-energy-selected prompt ions and gives a PIE curve,yielding ionization energy(Ip)with an uncertainty of 10-20 cm-1.In contrast,the MATI experiment detects threshold ions formed during the pulsed field ionization(PFI)process and gives a sharp peak at the ionization limit.When the sum of two photon energies is very close to the ionization threshold,both the prompt ions and the high n Rydberg neutrals are produced simultaneously in the“field-free”laser-molecular beam interaction zone(region I of our TOF lens system).These two species move with same velocity as the molecular beam of about 1500 m·s-1,depending on the carrier gas and stagnation pressure in the pulsed nozzle.22A pulsed electric field of-1.0 V·cm-1(duration=10 μs)(-2.5 V at TOF plate U1;0 V at plate U2;distance of plate U1 and U2 is 2.5 cm)is switched on about 20 ns after the occurrence of the laser pulses to guide the prompt ions towards the opposite direction of the ion detector.Because the Rydberg neutrals are not affected by the electric field,they keep moving to enter the field ionization region.After a time delay of more than 8.0 μs,the Rydberg neutrals reach the field-ionization zone(region II of our TOF lens system).It follows that a pulsed field of 200 V·cm-1(duration=10 μs)(+2250 V at TOF plate U2;+2050 V at plate U3;0 V at plate U4;distance between any two of these three plates is 1 cm)is applied in region II to field-ionize the high n Rydberg neutrals and to accelerate the newly formed threshold ions toward the dualstacked microchannel plate(MCP)ion detector.As stated above,the first pulsed electric field of-1.0 V·cm-1is referred as the spoiling field which rejects the prompt ions and causes the measured value to be lower than the true adiabatic Ipdue to the Stark effect.24,25The energy lowering(in cm-1)due to the electrical field effect may be estimated by 4.0F1/2when a pulsed field F(in V·cm-1)is applied.Under our experimental conditions,F is-1 V·cm-1.This means that the measured value is lowered than the true Ipby 4 cm-1.26
The signal from the MCP ion detector is accumulated and analyzed by a multichannel scaler(MCS,Stanford Research Systems,SR430).Each TOF mass spectrum at a particular laser wavelength is accumulated for 300 laser shots to avoid spurious signals due to shot-to-shot laser fluctuation and shown on the screen of the MCS.The MCS and a transient digitizer which monitors laser pulses are interfaced to a personal computer(PC).All TOF mass spectra are saved in the PC at a 0.020 nm(equivalent to 1.2 cm-1)interval over the entire scanning range of laser wavelength.Composite optical spectra of intensity versus wavelength are then constructed from the individual mass spectra.As the ion signal is proportional to the photon intensities of the excitation and ionization lasers in a two-color two-photon process,the obtained optical spectra are normalized to the laser power.
2.2 Computational method
We performed the ab initio and DFT calculations by using Gaussian 09 program package27to predict the optimized structures,total energies,vibrational frequencies,and other molecular properties of m-methylanisole in the S0,S1,and D0states.In the DFT calculations,both the Becke three-parameter functional with the Lee-Yang-Parr functional(B3LYP)and the PW91 functional(B3PW91)calculations with the 6-311++G(d,p)basis set can predict the electronic excitation and ionization energies with an uncertainty of no more than 5%.However,the latter gives a slightly better prediction on the vibrational frequencies in the S1and D0states.The configuration interaction singles(CIS),the timedependent(TD)B3PW91 calculations with the 6-311++G(d,p)basis set were applied to predict the vibrational frequencies of the S1state.When scaling factors of 0.93 and 0.98 were applied to correct the combined errors stemming from basis set incompleteness,the negligence of electron correlation and vibrational anharmonicity,the calculated vibrational frequencies match well with the measured values from the vibronic spectroscopic experiments.The vibrational frequencies of the D0state were calculated at the unrestricted Hartree-Fock(HF)and B3PW91 levels with the 6-311++G(d,p)basis set.When scaling factors of 0.92 and 0.98 for cis-m-methylanisole and 0.90 and 0.96 for trans-mmethylanisole were applied,the calculated values became very close to the measured frequencies.The adiabatic Ipwas deduced from the difference in the zero-point energy(ZPE)levels of the cation in the D0state and the corresponding neutral species in the S0state.
3 Results
3.1 1C-R2PI spectrum of m-methylanisole
Fig.2 shows the 1C-R2PI spectrum of m-methylanisole over an energy range of 1600 cm-1,recorded by using the 1C-R2PI technique.The respective band origins of the S1←S0electronic excitation(E1)of the cis and trans rotamers appear at(36049±2)and(36117±2)cm-1,which are in excellent agreement with those reported previously.5-8In the present experimental conditions,the full width at half maximum(FWHM)of these bands is 5-7 cm-1.Therefore,the origin bands may contain the methyl internal rotational levels 0a1and 1e.
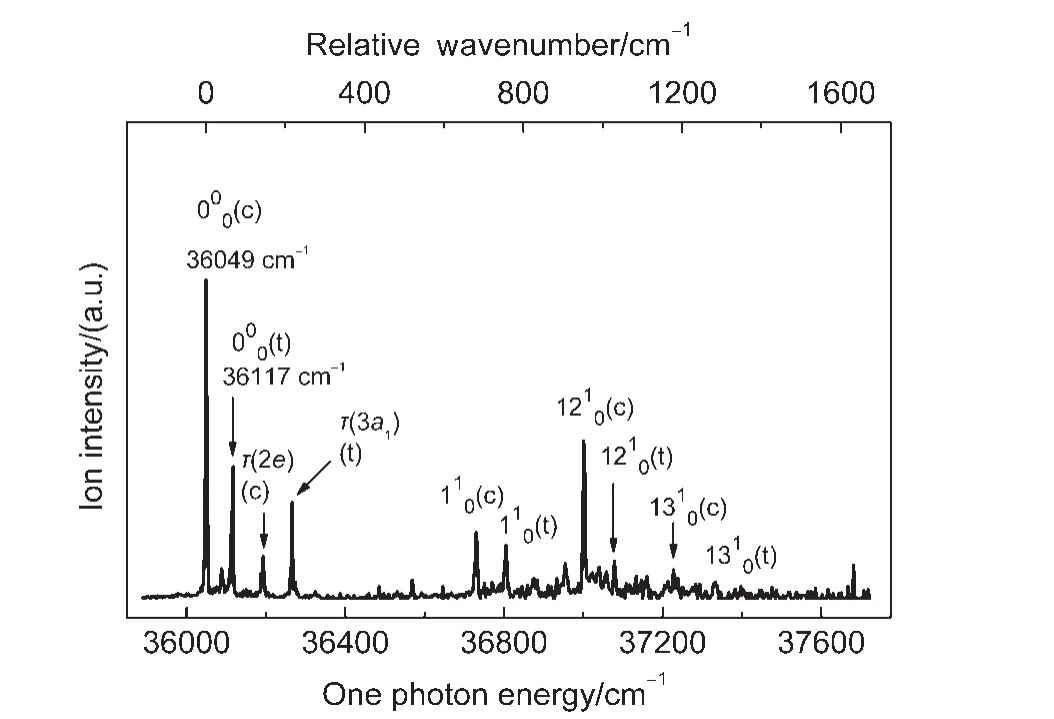
Fig.2 1C-R2PI spectrum of m-methylanisole near the S1←S0electronic transition
The observed vibronic bands along with the excitation photon energy,relative intensity,energy shift from the band origin,cal-culated vibrational frequency,and possible assignment are listed in Table 1.We use“τ”to designate the CH3torsion throughout this paper.The spectral assignment has been achieved by comparing the present data with those in the literature5-9,28and the predicted values from the CIS and TD-B3PW91 calculations with the 6-311++G(d,p)basis set.The numbering system of the normal vibrations follows that used by Varsanyi and Szoke29for benzene derivatives and is based on Wilson′s notations.30According to Varsanyi,28m-methylanisole is classified as an m-Di-“light”substituted benzene.These normal vibrations can be viewed by following the GaussView procedure of the Gaussian 09 program.27Previous studies5-9are mainly on the spectral bands related to the CH3torsion.Here,we focus on those resulting from the in-plane ring and substituent-sensitive vibrations.
m-Methylanisole has 51 normal vibrations including 30 benzene-like,9 methyl,and 12 methoxyl modes.Only those active vibrations in the S1state with large Franck-Condon overlaps can be observed in the 1C-R2PI spectrum.Because the vibration frequency of each mode of multiply substituted benzene is related to substituent-ring and substituent-substituent interactions,each molecule has a unique set of spectral features.Therefore,a wellresolved spectrum can be used for molecular identification,although the spectral assignment is not trivial.The bands at 36193 and 36266 cm-1in Fig.2 corresponding to shifts by 144 and 149 cm-1on the high energy side of the band origins of the two respective rotamers result from the CH3torsion τ(2e)of cis-mmethylanisole and τ(3a1)of trans-m-methylanisole.This finding is consistent with that reported in the literature.6,9The assignment for the observed vibronic transitions related to the ring vibrations of the rotamers of m-methylanisole has been achieved by comparing the present experimental data with those of the corresponding rotamers m-fluoroanisole12and m-chloroanisole31as well as the calculated results.The vibronic transitions related to vibrations 6b1,11,7b1,121,and 131are found to have frequencies of 519,681,827,952,and 1178 cm-1for cis-m-methylanisole and 529,688,838,961,and 1215 cm-1for trans-m-methylanisole.Modes 6b,1,and 12 mainly involve in-plane ring deformation,whereas modes 7b and 13 designate ring C1―OCH3and ring C3―CH3stretching vibrations,respectively.In-plane ring deformation 6a of the cis rotamer appears at 437 cm-1with low intensity.It has been reported that mode 1 has frequency of 693 cm-1for both cisand trans-m-fluoroanisole12and 626 and 639 cm-1for cis-and trans-m-chloroanisole,31respectively.These data show that the frequency difference of each normal vibration of the two rotamers depends on the nature,vibrational pattern,and relative orientation of the substituents.
3.2 Cation spectra of selected cis-and transm-methylanisole
To investigate molecular properties of selected rotamers of mmethylanisole,we have performed the PIE and MATI experiments.When the sum of the photon energies of the pump and probe laser is close to the ionization limit,both the high n Rydberg neutrals and prompt ion are produced simultaneously in the“field free”laser-molecular beam interaction region(region I of our TOF lens system).22After a time delay of about 11.2 μs,the Rydberg molecules are field ionized along with the prompt ions and accelerated in region II of our TOF lens system.22This leads to an abrupt change of the total ion current near the ionization limit in a PIE spectrum.Fig.3 shows the PIE curves of cis-and trans-mmethylanisole,recorded by ionizing via their respective S100levels at 36049 and 36117 cm-1.The first rising step in the PIE of cis-mmethylanisole indicates that the Ipis(64859±10)cm-1.Two more steps clearly appear at 64957 and 65043 cm-1,which are higher than the first one by 98 and 184 cm-1,respectively.They corre-spond to the internal methyl rotations τ(2e)and τ(3a1)of the cism-methylanisole cation.Similarly,the Ipof trans-m-methylanisole is found to be(65110±10)cm-1.Two steps near 65199 and 65262 cm-1result from the CH3torsion τ(2e)and τ(3a1)of the trans-mmethylanisole cation with the respective frequencies of 89 and 152 cm-1.When we performed the MATI experiments which detect only the threshold ions,we observed sharp peaks corresponding to these abruptly rising steps,as seen in Figs.4 and 5.A similar observation of the out-of-plane ring deformation vibration of the benzimidazole32cation has been reported.

Table 1 Observed bands in the 1C-R2PI spectrum of m-methylanisole in Fig.2
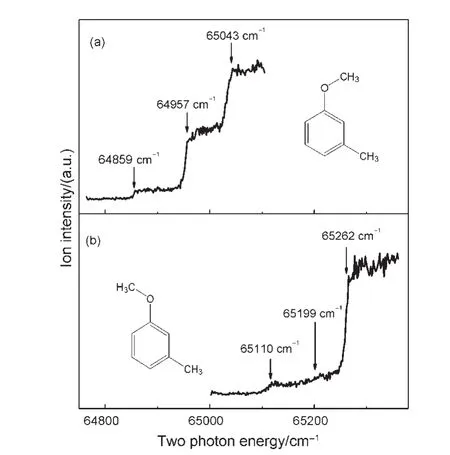
Fig.3 PIE curves of(a)cis-m-methylanisole and(b)trans-mmethylanisole,recorded by ionizing via their respective S100 levels
Fig.4 shows the MATI spectra of cis-m-methylanisole,recorded by ionizing via the 00(36049 cm-1),τ(2e)(00+144 cm-1),and 6b1(00+519 cm-1)levels in the S1state.Although some portions have low intensity,the signal-to-noise ratio of these spectra is reasonably good.Consideration of the uncertainty of the laser photon energy,the spectral width,and the Stark effect,the adiabatic Ipof cis-m-methylanisole is determined to be(64859±5)cm-1((8.0415±0.0006)eV),which is in good agreement with that measured by our PIE method and by the ZEKE experiments reported by Fujii and coworkers9.The observed MATI bands resulting from the cation vibrations and their possible assignments are listed in Table 2.As stated previously,the band origin may include the methyl internal rotational levels 0a1and 1e.When the S100state is used as the intermediate level,the pronounced bands at 98,180,248,and 307 cm-1correspond to the CH3torsion τ(2e),τ(3a1),τ(5e),and τ(6a1)of cis-m-methylanisole cation,as reported in the literature.9The weak bands at 648,733,798,and 857 cm-1are caused by the combinational motion of the in-plane ring deformation 6b1vibration and the CH3torsion τ(2e), τ(3a1), τ(5e),and τ(6a1),respectively.Another weak band at 951 cm-1results from the inplane ring deformation 121vibration.When the S1τ(2e)state is used as the intermediate level,the pronounced bands at 249,338,and 416 cm-1correspond to the CH3torsion τ(5e),τ(7e),and τ(8e).This assignment fulfills the selection rule of e↔e transition.The weak bands at 803 and 958 cm-1correspond to the combinational motion 6b1τ(5e)and the in-plane ring deformation 121vibration.When the MATI spectrum is recorded by ionizing via the vibronic S16b1state which may contain CH3torsion with the a and e symmetry,the weak bands at 180 and 250 cm-1are assigned to the CH3torsion of τ(3a1)and τ(5e).9The distinct bands at 648,733,796,and 857 cm-1correspond to the combinational motion of vibration 6b1and the CH3torsion of τ(2e),τ(3a1),τ(5e),and τ(6a1).The weak bands at 549 and 1280 cm-1result from vibrations 6b1and 131,respectively.
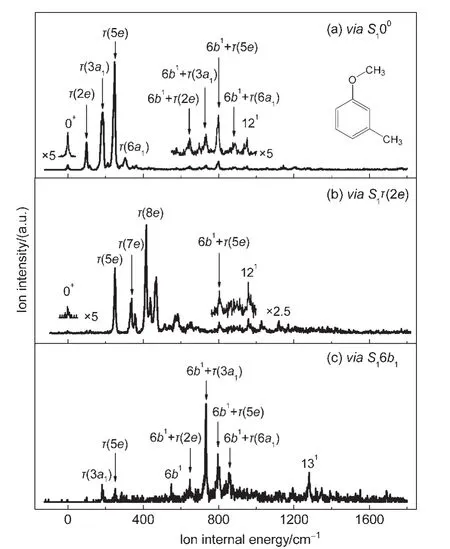
Fig.4 MATI spectra of cis-m-methylanisole,recorded by ionizing via the(a)S100,(b)S1τ(2e),and(c)S16b1 intermediate states
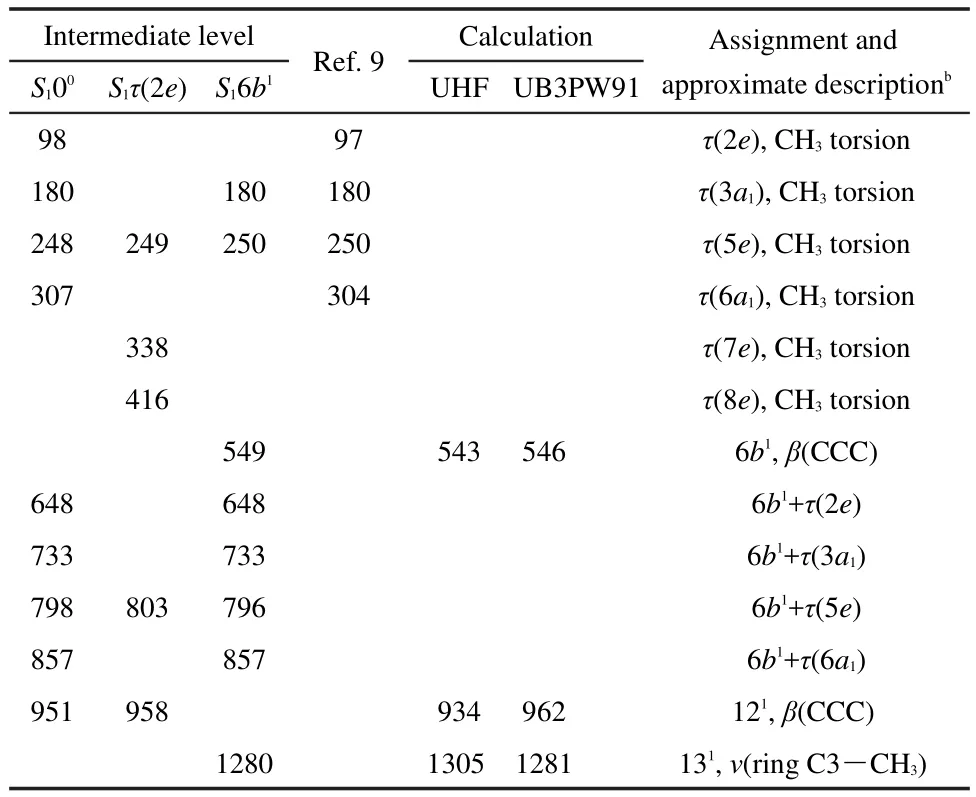
Table 2 Observed bands(cm-1)in the MATI spectra of cis-mmethylanisole in Fig.4a

Fig.5 MATI spectra of trans-m-methylanisole,recorded by ionizing via the(a)S100,(b)S1τ(3a1),and(c)S16b1 intermediate states
Fig.5 shows the MATI spectra of trans-m-methylanisole,recorded by ionizing via the 00(36117 cm-1),τ(3a1)(00+149 cm-1),and 6b1(00+529 cm-1)levels in the S1state.The adiabatic Ipis determined to be(65110±5)cm-1((8.0726±0.0006)eV)which is also in excellent agreement with that measured by our PIE method and by the ZEKE experiments.9The observed MATI bands and their possible assignments are listed in Table 3.As seen in Fig.5(a),when the S100state is used as the intermediate state,the lowfrequency bands at 89,152,and 268 cm-1result from the CH3torsion τ(2e),τ(3a1),and τ(6a1)of the trans-m-methylanisole cation,as reported previously.9The weak bands at 600 and 709 cm-1correspond to the combinational motions 151τ(3a1)and 6b1τ(3a1),respectively.Another weak band at 860 cm-1is tentatively assigned to the substituent-sensitive ring C1―OCH3stretching vibration 7b1.It is interesting to note that when the S1τ(3a1)is used as the intermediate level,many weak MATI bands related to inplane ring fundamental vibrations of the cation are observed,as seen in Fig.5(b).The bands at 446,555,959,1085,and 1160 cm-1are related to vibrations 151,6b1,121,18a1,and 9b1,respectively.The band at 708 cm-1corresponds to the combinational motion6b1τ(3a1).When the MATI spectrum is recorded by ionizing via the S16b1vibronic level,distinct bands at 712 and 823 cm-1correspond to the combinational motions 6b1τ(3a1)and 6b1τ(6a1)of the trans-m-methylanisole cation,respectively.The weak bands at 600 and 1269 cm-1result from the combinational motion 151τ(3a1)and the substituent-sensitive ring C3―CH3stretching vibration 131,respectively.
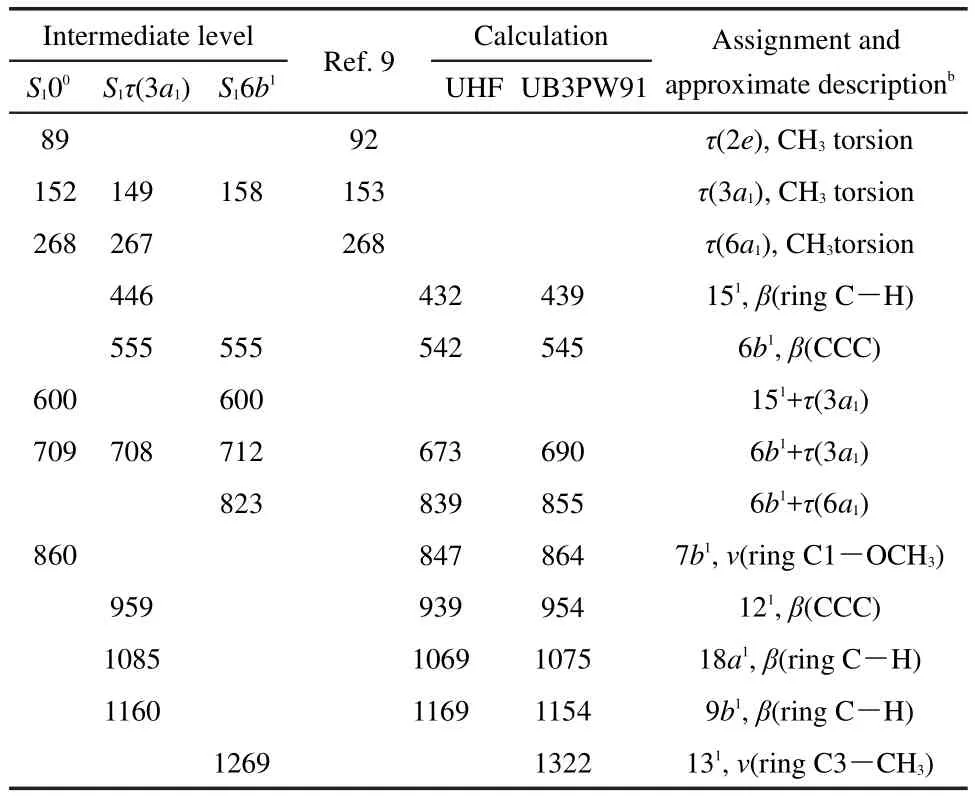
Table 3 Observed bands(cm-1)in the MATI spectra of trans-mmethylanisole in Fig.5 a
4 Discussion
4.1 Optimized structures of m-methylanisole in the S0,S1,and D0states
Our theoretical calculations predict that in the S0,S1,and D0states m-methylanisole has stable cis and trans configurations(Fig.1)as in the cases of m-fluoroanisole12and m-chloroanisole31.Fig.4(a)and Fig.5(a)show a very weak 0+band of the MATI spectrum recorded by ionizing via the S100intermediate levels of cis-and trans-m-methylanisole.This observation is distinctly different from that of m-fluoroanisole and m-chloroanisole.Previous studies show that molecular geometry and some in-plane vibrations in the D0state resemble those in the S1state for both rotamers of m-fluoroanisole12and m-chloroanisole.31In contrast,the present results may indicate that the geometry of the cis and trans rotamers of m-methylanisole changes drastically upon the D0←S1transition.Here,we present the results from the theoretical calculations to support the experimental findings which imply that interaction of the CH3substituent with the aromatic ring is quite different from that of either F or Cl atom.
Table 4 lists the optimized structural parameters of cis-and trans-m-methylanisole in the S0,S1,and D0states,predicted by the restricted B3PW91,TD-B3PW91,and unrestricted B3PW91 calculations with the 6-311++G(d,p)basis set.It is clear that the substituent-ring interaction causes the aromatic ring to distort from a regular hexagon.It follows that the bond lengths and bond angles of cis-and trans-m-methylanisole change upon electronic transition.As shown in Table 4,the S1←S0excitation leads to an expansion of the aromatic ring for both rotamers.Here,we use the cis rotamer as an example for discussion.The electronic excitation causes the lengths of the C1―C2,C2―C3,C3―C4,C4―C5,C5―C6,and C6―C1 bonds of cis-m-methylanisole(see Fig.1)to increase from 0.1393,0.1401,0.1391,0.1396,0.1383,and 0.1399 nm to 0.1420,0.1427,0.1420,0.1411,0.1419,and 0.1418 nm,respectively.It is noted that the C1―OCH3and C3―CH3bonds slightly shrink from 0.1361 to 0.1345 nm and 0.1505 to 0.1488 nm,respectively.The shortening of these two substituent related bonds indicates that the interaction of the CH3and OCH3groups with the aromatic ring becomes slightly enhances upon S1←S0transition.The change of bond angles ∠C6C1C2,∠C2C3C4,and ∠C3C4C5 from 120.0º,119.1º,120.2ºto 123.2º,118.3º,and 123.2ºalso implies that the substituent-ring interaction varies upon the electronic transition.
The D0←S1transition involves removing one electron from cism-methylanisole in the S1state.The data in Table 4 showed that the lengths of the C2―C3,C5―C6,and C1―OCH3bonds decrease substantially from 0.1427,0.1419,and 0.1345 nm to 0.1376,0.1372,and 0.1304 nm,respectively.This implies that the D0←S1transition leads to double bond characters in the C2―C3,C5―C6,and C1―OCH3bonds.In consideration of the structure in Fig.1,the ring part of cis-m-methylanisole in the cationic D0state is similar to that of quinone.4These results show that the D0←S1transition leads to a significant change in molecular geometry.The predicted structural change of trans-m-methylanisole uponthe S1←S0and D0←S1transitions is similar to that of cis-mmethylanisole,as shown in Table 4.The B3PW91/6-311++G(d,p)calculations predict that the aromatic ring of both rotamers of mmethylanisole expands upon the S1←S0electronic excitation.Furthermore,the ring part changes significantly from a distorted hexagon to a quinone-like structure in the D0←S1transition.These calculated results support the experimental findings of the very weak 0+bands in the MATI spectra of cis-and trans-m-methylanisole,as seen in Fig.4(a)and Fig.5(a).

Table 4 Bond lengths and angles of cis-and trans-mmethylanisole in the S0,S1and D0states,predicted by the restricted B3PW91,TD-B3PW91,and unrestricted B3PW91 methods with the 6-311++G(d,p)basis set,respectively
4.2 Configuration and substitution effects on the E1and I p
The respective E1of cis-and trans-m-methylanisole are measured to be 36049 and 36117 cm-1.The CIS/6-311++G(d,p)calculations predict these values to be 45173 and 45335 cm-1.Moreover,the TD-B3LYP and TD-B3PW91 methods with the same basis set give 37341 and 37717 cm-1for the cis rotamer,and 37371 and 37796 cm-1for the trans rotamer,respectively.These results show that the CIS,TD-B3LYP,and TD-B3PW91 methods overestimate the E1by about 25%,3.5%,and 4.6%,respectively.The adiabatic Ipof the cis and trans rotamers are determined to be 64859 and 65110 cm-1,respectively.9The HF/6-311++G(d,p)calculations predict these values to be 52968 and 53278 cm-1,which are less than the measured values by about 18%.The B3LYP and B3PW91 methods with the same basis set give 63089 and 63263 cm-1for the cis rotamer,and 63415 and 63601 cm-1for the trans rotamer.These results show that the B3LYP and B3PW91 calculated values are less than the measured ones by about 2.7%and 2.4%,respectively.Evidently,these calculations show that the DFT method is better than the HF method in predicting the E1and Ipof both rotamers.In addition,all of these calculations predict that the cis rotamer has a slightly smaller E1and Ipthan the trans rotamer of m-methylanisole.Similar findings have been reported for m-fluoroanisole12and m-chloroanisole.31
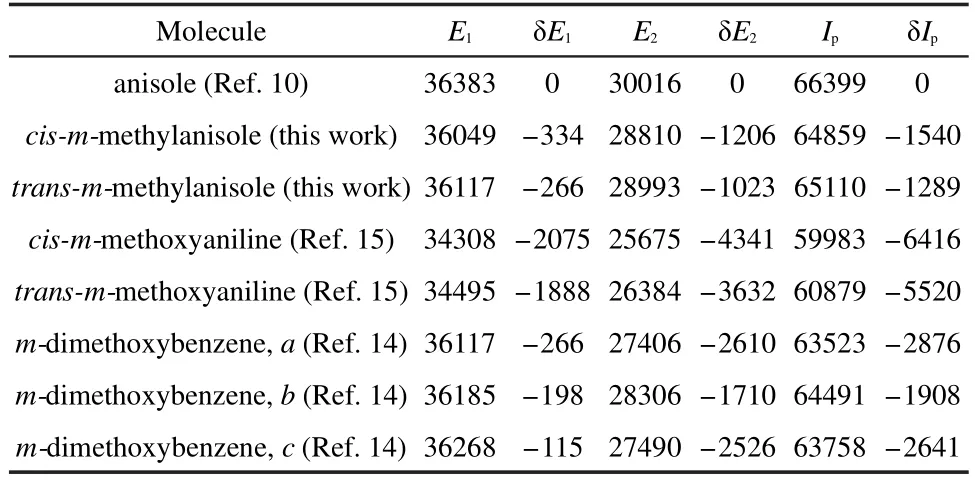
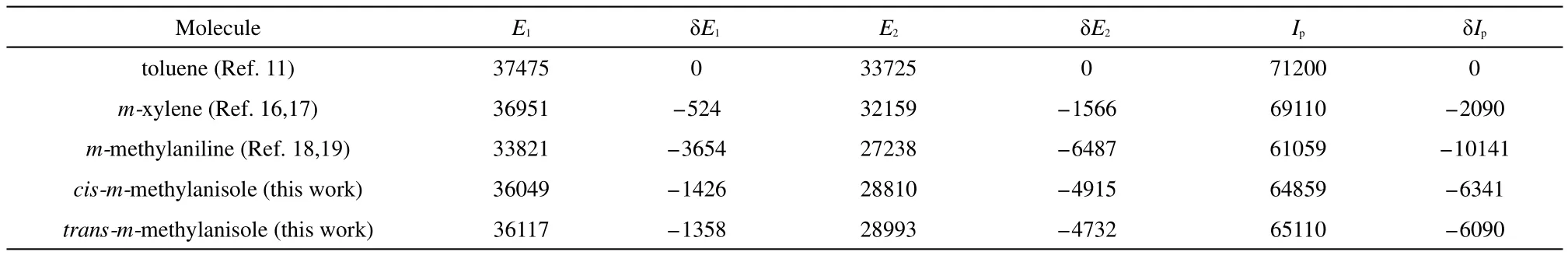
The experimental and calculated results show that the cis is more stable than the trans rotamer in the upper(S1and D0)states than the lower(S0)state.This indicates that the through-space(or roaming)interaction between the CH3and OCH3groups of the cis rotamer is stronger than that of the trans rotamer in the upper state.The difference in the E1and Ipof the two rotamers of mmethylanisole results from different degrees of interaction between the two substituents in various energy states.Similar findings have been reported for m-cresol,3,20m-fluoroanisole,3,12mchloroanisole,31m-fluorophenol,3,33m-chlorophenol,3m-methoxyaniline,15m-aminophenol,34,353,4-difluoroanisole,36and 3,4-difluorophenol.37
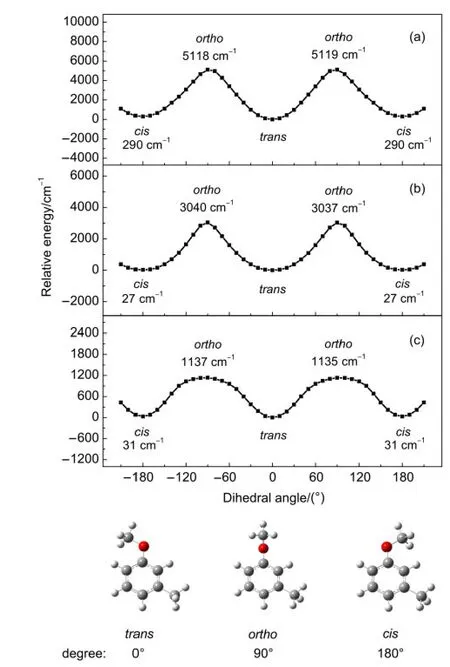
Our restricted B3PW91/6-311++G(d,p)calculations predict that in the S0state the ZPE level of cis-m-methylanisole is higher than that of trans-m-methylanisole by 38 cm-1.With the measured values stated previously,the ZPE level in the S1state of the cis rotamer is calculated to be lower than that of the trans rotamer by 106 cm-1,as shown in Fig.6.Similarly,the adiabatic Ipof the cis and trans rotamers of m-methylanisole are determined to be 64859 and 65110 cm-1by our MATI experiments,respectively.The ZPE level in the cationic D0state of the cis rotamer is then deduced to be lower than that of the trans rotamer by 289 cm-1.These experimental data and calculated results imply that the throughspace interaction between the CH3and OCH3substituents at the meta position of the cis rotamer is stronger than that of the trans rotamer of m-methylanisole.Therefore,the strength of the throughspace substituent-substituent interaction of m-methylanisole in the electronic state follows the order:S0 It is known that a substituent can interact with an aromatic ring by inductive effect through the σ bond and by the resonant(or mesomeric)effect through the π bond.4The inductive effect,which is related to the electronegativity of the substituent,reflects the ability of donating or accepting electrons from the substituent to the ring.The resonant effect depends on the extent of π orbital overlap of the substituent with the ring.In addition,the hydrogen atoms of an alkyl group can interact with an aromatic group by hyperconjugation.The collective effect of the above mentioned factors can cause a slight change in the nearby electron density of the aromatic ring,leading to a lowering of the ZPE level of the electronic state.If the degree of the lowering of the ZPE level of the upper state is greater than that of the lower one,it gives smaller transition energy.Oppositely,it yields a slightly larger transition energy.26 Fig.6 Energy level diagram of the cis and trans rotamers of mmethylanisole in the S0,S1,and D0states Table 5 lists the measured E1and Ipof anisole and its derivative,on the basis of the LIF,REMPI,ZEKE,and MATI experiments.10,14,15The excitation energy of the D0←S1transition(E2)are also shown in Table 5.These anisole derivatives have a second substituent of CH3,NH2,and OCH3at the meta position with respect to the OCH3group.Previous studies10,14,15show that both m-methylanisole and m-methylaniline have two rotamers,whereas m-dimethoxybenzene has three rotamers involved in the two-photon excitation and ionization processes.As shown in Table 5,the E1and Ipof mmethylanisole,m-methoxyaniline,and m-dimethoxybenzene are less than those of anisole.The observed smaller transition energy of the anisole derivatives indicates that the interaction of the CH3,NH2,and OCH3groups and the aromatic ring is stronger in the upper(i.e.S1or D0)state than that in the lower(i.e.S0)state.Table 6 lists the measured E1and Ipof toluene11and its derivatives.16-19It is evident that substitution of the CH3,NH2,and OCH3groups leads to a smaller E1and Ip.These data support the previous statement that the substituent-ring interaction of these anisole and toluene derivatives is stronger in the upper electronically excited S1(or cationic D0)state than that in the lower ground S0state. Figs.2,4,and 5 show that some fundamental in-plane ring and substituent-sensitive vibrations are active in addition to the CH3torsion for both the cis and trans rotamers of m-methylanisole in the S1and D0states.As listed in Table 7,frequencies of modes 6b1,11,7b1,121,and 131are measured to be 519,681,827,952,and 1178 cm-1for cis-m-methylanisole and 529,688,838,961,and 1215 cm-1for trans-m-methylanisole in the S1state.The vibrational frequencies of these modes of the cis rotamer are slightly less than those of the trans rotamer.Furthermore,frequencies of vibrations 6b1,121,and 131are measured to be 549,951,and 1280 cm-1for cis-m-methylanisole and 555,959,and 1269 cm-1for trans-m-methylanisole in the D0state.This indicates that frequencyis slightly different for each mode of the two rotamers.In consideration of the molecular geometry changes upon the D0←S1transition,we propose that the nature,relative orientation of the CH3and OCH3substituents,molecular geometry,and vibrational pattern can influence the frequency of normal modes. Table 5 Measured transition energies(cm-1)of anisole and its derivatives Table 6 Measured transition energies(cm-1)of toluene and its derivatives Table 7 Measured frequencies(cm-1)of active vibrations of m-methylanisole in the S1and D0states Fig.7 One-dimensional potential energy surfaces for the rotation of the C―OCH3bond of m-methylanisole in the(a)D0,(b)S1,and(c)S0states As discussed previously,the cis and trans rotamers of mmethylanisole involve in two-photon excitation and ionization processes.We have performed the restricted B3PW91,TDB3PW91,and unrestricted B3PW91 calculations with the 6-311++G(d,p)basis set to investigate the cis-trans isomerization of mmethylanisole in the S0,S1,and D0states,respectively.It was achieved by scanning the one-dimensional potential energy surface on the rotation of the C1―OCH3.As illustrated in Fig.7(c),in the S0state,the trans rotamer has the lowest total electronic energy,whereas the cis configuration lies in an energy level higher by 31 cm-1.In addition,an intermediate species with the orthogonal form(with the C1―O―CH3angle of 90°)has energy higher than that of the trans con?guration by 1136 cm-1.This result predicts that the trans form is the most stable configuration and the energy barrier of the cis-trans isomerization is in the range of 1105-1136 cm-1.Similarly,the most stable structure of mmethylanisole in the S1and D0states also has the trans configuration,as shown in Fig.7(b)and 7(a).However,the isomerization barriers are greatly enhanced to 3011-3038 cm-1and 4828-5118 cm-1in the S1and D0states,respectively.The increase of isomerization barriers is due to the fact that the interaction between the OCH3group and the ring is stronger in the upper(S1or D0)state than that in the lower S0state.As listed in Table 4,these DFT calculations predict the C1―OCH3bond lengths to be 0.1361,0.1345,and 0.1304 nm for cis-m-methylanisole and 0.1361,0.1344,and 0.1302 nm for trans-m-methylanisole in the S0,S1,and D0states,respectively.A shorter length implies a stronger chemical bond.Therefore,the isomerization energy barrier to the C1―OCH3rotation follows the order:S0 We have applied the 1C-R2PI,2C-R2PI,and MATI techniques to record the 1C-R2PI,PIE,and cation spectra of m-methylanisole.The E1of the cis and trans rotamers are determined to be 36049 and 36117 cm-1,whereas the adiabatic Ipare 64859 and 65110 cm-1,respectively.Our restricted B3PW91/6-311++G(d,p)calculations predict that in the S0state the ZPE level of cis-mmethylanisole is higher than that of trans-m-methylanisole by 38 cm-1.With the measured E1and Ip,the ZPE levels of the cis rotamer in the S1and D0states are calculated to be lower than those of the trans rotamer by 106 and 289 cm-1,respectively.These imply that the through-space(or“roaming”)interaction between the CH3and OCH3substituents of m-methylanisole in the electronic state follows the order:S0 Analysis of the 1C-R2PI and cation spectra show that several in-plane ring and substituent-sensitive vibrations are active in addition to the CH3torsion for both the cis and trans rotamers of m-methylanisole in the S1and D0states.However,the frequency difference of each vibration may depend on the nature,vibrational pattern,relative orientation,and the extent of the substituents participated in the normal vibration. (1)Dian,B.C.;Longarte,A.;Winter,P.R.;Zwier,T.S.J.Chem.Phys.2004,120,133.doi:10.1063/1.1626540 (2)Pan,C.P.;Barkley,M.D.Biophys.J.2004,86,3828.doi:10.1529/biophysj.103.038901 (3)Oikawa,A.;Abe,H.;Mikami,N.;Ito,M.Chem.Phys.Lett.1985,116,50.doi:10.1016/0009-2614(85)80123-8 (4)McMurry,J.Organic Chemistry,7th ed.;Brooke/Cole Publishing Company:Belmont,California,2007. (5)Breen,P.J.;Bernstein,E.R.;Secor,H.V.;Seeman,J.I.J.Am.Chem.Soc.1989,111,1958.doi:10.1021/ja00188a002 (6)Ichimura,T.;Suzuki,T.J.Photochem.Photobiol.C:Photochem.Rev.2000,1,79.doi:10.1016/S1389-5567(00)00006-X (7)Kojima,H.;Miyake,K.;Sakeda,K.;Suzuki,T.;Ichimura,T.;Tanaka,N.;Negishi,D.;Takayanagi,M.;Hanazaki,I.J.Mol.Struct.2003,655,185.doi:10.1016/S0022-2860(03)00258-8 (8)Alvarez-Valtierra,L.;Yi,J.T.;Pratt,D.W.J.Phys.Chem.B 2006,110,19914.doi:10.1021/jp062050h (9)Kinoshita,S.;Kojima,H.;Suzuki,T.;Ichimura,T.;Yoshida,K.;Sakai,M.;Fujii,M.Phys.Chem.Chem.Phys.2001,3,4889.doi:10.1039/b105719g (10)Pradhan,M.;Li,C.;Lin,J.L.;Tzeng,W.B.Chem.Phys.Lett.2005,407,100.doi:10.1016/j.cplett.2005.03.068 (11)Lu,K.T.;Eiden,G.C.;Weisshaar,J.C.J.Phys.Chem.1992,96,9742.doi:10.1021/j100203a032 (12)Shiung,K.S.;Yu,D.;Huang,H.C.;Tzeng,W.B.J.Mol.Spectrosc.2012,274,43.doi:10.1016/j.jms.2012.04.004 (13)Ullrich,S.;Geppert,W.D.;Dessent,C.E.H.;Müller-Dethlefs,K.J.Phys.Chem.A 2000,104,11864. (14)Yang,S.C.;Huang,S.W.;Tzeng,W.B.J.Phys.Chem.A 2010,114,11144.doi:10.1021/jp1026652 (15)Lin,J.L.;Huang,C.J.;Lin,C.H.;Tzeng,W.B.J.Mol.Spectrosc.2007,244,1.doi:10.1016/j.jms.2007.05.001 (16)Zhang,S.;Tang,B.F.;Wang,Y.M.;Zhang,B.Chem.Phys.Lett.2004,397,495.doi:10.1016/j.cplett.2004.09.025 (17)Held,A.;Selzle,H.L.;Schlag,E.W.J.Phys.Chem.A 1998,102,9625. (18)Lin,J.L.;Lin,K.C.;Tzeng,W.B.J.Phys.Chem.A 2002,106,6462.doi:10.1021/jp0204713 (19)Okuyama,K.;Mikami,N.;Ito,M.Laser Chem.1987,7,197.doi:10.1155/LC.7.197 (20)Huang,J.;Huang,K.;Liu,S.;Luo,Q.;Tzeng,W.B.J.Photochem.Photobiol.A:Chem.2007,188,252.doi:10.1016/j.jphotochem.2006.12.016 (21)Huang,J.;Lin,J.L.;Tzeng,W.B.Spectrochim.Acta A 2007,67,989.doi:10.1016/j.saa.2006.09.018 (22)Tzeng,W.B.;Lin,J.L.J.Phys.Chem.A 1999,103,8612. (23)Sinclair,W.E.;Pratt,D.W.J.Chem.Phys.1996,105,7942.doi:10.1063/1.472710 (25)Schlag,E.W.ZEKE Spectroscopy;Cambridge University Press:Cambridge,1998;pp 40-41. (26)Zhang,B.;Li,C.;Su,H.;Lin,J.L.;Tzeng,W.B.Chem.Phys.Lett.2004,390,65.doi:10.1016/j.cplett.2004.04.013 (27)Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09,Revision A.02;Gaussian Inc.:Wallingford,CT,2009. (28)Varsanyi,G.Assignments of Vibrational Spectra of Seven Hundred Benzene Derivatives;Wiley:New York,1974;pp 185,201. (29)Varsanyi G.;Szoke,S.Vibrational Spectra of Benzene Derivatives;Academic Press:New York,London,1969. (30)Wilson,E.B.Phys.Rev.1934,45,706.doi:10.1103/PhysRev.45.706 (31)Huang,H.C.;Shiung,K.S.;Jin,B.Y.;Tzeng,W.B.Chem.Phys.2013,425,114.doi:10.1016/j.chemphys.2013.08.013 (32)Lin,J.L.;Li,Y.C.;Tzeng,W.B.Chem.Phys.2007,334,189.doi:10.1016/j.chemphys.2007.03.002 (33)Yosida,K.;Suzuki,K.;Ishiuchi,S.;Sakai,M.;Fujii,M.;Dessent,C.E.H.;Müller-Dethlefs,K.Phys.Chem.Chem.Phys.2002,4,2534.doi:10.1039/b201107g (34)Shinozaki,M.;Sakai,M.;Yamaguchi,S.;Fujioka,T.;Fujii,M.Phys.Chem.Chem.Phys.2003,5,5044.doi:10.1039/b309461h (35)Xie,Y.;Su,H.;Tzeng,W.B.Chem.Phys.Lett.2004,394,182.doi:10.1016/j.cplett.2004.07.005 (36)Xu,Y.;Tzeng,S.Y.;Tzeng,W.B.Spectrochim.Acta A 2013,102,365.doi:10.1016/j.saa.2012.10.020 (37)Tsai,C.Y.;Tzeng,W.B.J.Photochem.Photobiol.A 2013,270,53.doi:10.1016/j.jphotochem.2013.07.014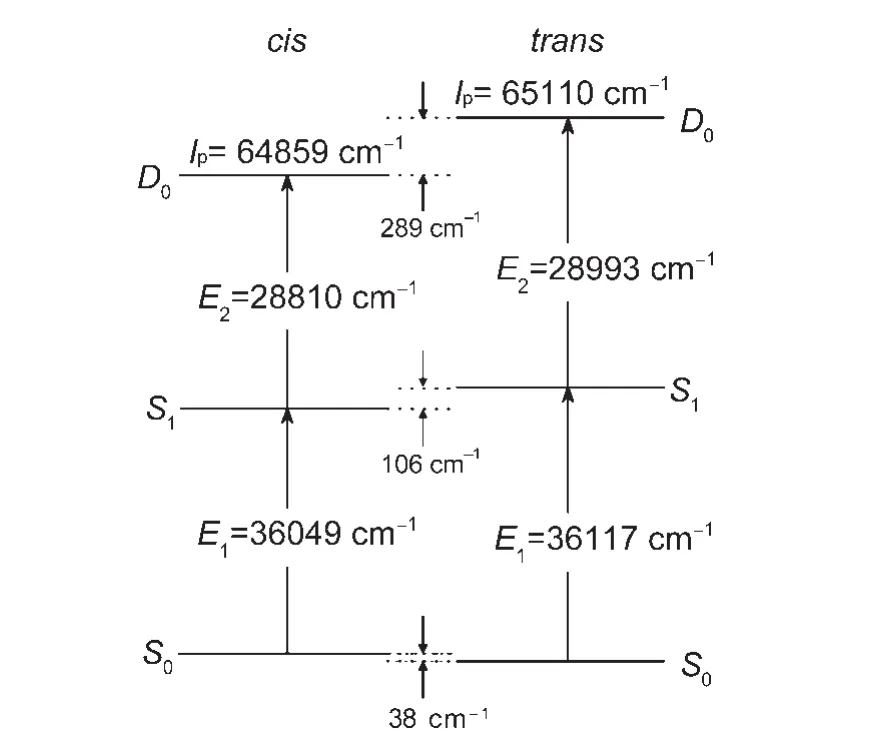
4.3 Frequencies of observed active vibrations in the S1and D0states

4.4 Energy barrier of the cis-trans isomerization of mmethylanisole
5 Conclusions
猜你喜欢
体育科技文献通报(2022年4期)2022-10-21现代装饰(2022年2期)2022-05-23少儿科学周刊·儿童版(2021年22期)2021-12-11少儿科学周刊·儿童版(2021年22期)2021-12-11少儿科学周刊·儿童版(2021年22期)2021-12-11现代装饰(2020年11期)2020-11-27军事文摘(2020年20期)2020-11-16小天使·三年级语数英综合(2018年9期)2018-10-16美与时代·美术学刊(2017年2期)2017-05-11美与时代·美术学刊(2017年2期)2017-05-11
