
螺桨烷型分子BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P)的结构和性质
2014-10-18 05:27李金霞张聪杰
物理化学学报 2014年3期
李金霞 张聪杰
(陕西师范大学化学化工学院,陕西省大分子科学重点实验室,西安 710062)
1 引言
[n.n.n]螺桨烷分子包含三个桥链和两个桥头C原子,其中桥头C原子具有“翻转”的四面体几何构型.[l.l.l]螺桨烷C5H61是最小的[n.n.n]螺桨烷,因为该分子桥头的两个C原子之间的化学键问题,使其长期在理论和实验上一直是研究热点.2-17最近通过价键理论、分子轨道理论以及分子中原子理论计算,结果表明[l.l.l]螺桨烷C5H6中桥头C―C化学键不是经典的共价键和离子键,而是共价键和离子键的混合键,称之为电荷转移(charge-shift)键.14-17而且,实验上也合成了[2.2.2]螺桨烷及其衍生物,18,19[4.1.1]螺桨烷,20,21[3.1.1]螺桨烷及其衍生物,22,23[3.2.l]螺桨烷24,25等一系列化合物.除了全碳型的螺桨烷外,含重原子的螺桨烷E5R6(E=Si,Ge,Sn)及其衍生物在实验和理论上也引起了广泛的关注.26-36例如,实验上合成了杂原子[1.1.1]螺桨烷Ge2Si3Mes6和Sn2Si3Mes6(Mes=2,4,6-Me3C6H2),33Ge2{Sn-(Cl)R}3(R=2,6-Mes2C6H3;Mes=2,4,6-Me3C6H2)34和含Ge原子的螺二(五角星[1.1.1]螺桨烷)35以及[1.1.1]2,4,5-硒化砷-1,3-二硅[1.1.1]戊烷.36除此之外,实验上也成功地合成了桥头原子非第IV主族原子的螺桨烷类似物(H2E)3Bi2.37
因为B―N与其等电子C―C键存在很多相似和不同的性质,38-40所以我们将[n.n.n]螺桨烷中的两个桥头C原子用B和N/P原子取代后得到了螺桨烷型分子BX[(CH2)n]3(X=N,P;n=1-6)化合物.而且,为了讨论桥链的二面角变化对[1.1.1]螺桨烷型分子的稳定性、化学键以及电子光谱等性质的影响,我们进一步研究了BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)的结构和性质.通过理论方法计算了该类化合物的结构、化学键和电子光谱性质,以期研究螺桨烷中桥头为不对称原子时的化学性质.
2 计算方法
采用密度泛函理论(DFT)41,42中的B3LYP43,44方法,所有原子采用6-311++G**基组,对[n.n.n]螺桨烷型分子BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)进行了结构优化,并且在相同水平上计算了振动频率.为了方便,我们将BN[(CH2)n]3和BP[(CH2)n]3(n=1-6)分别用A1-A6和B1-B6表示,相应的BN(CH2)[CH(CH2)nCH]和BP(CH2)[CH(CH2)nCH](n=1-6)分别用C1-C6和D1-D6表示.利用自然键轨道(NBO)法45计算了它们的Wiberg键级、电荷分布和电子组态结构.采用含时密度泛函理论(TDDFT)中TD-B3LYP46方法以及6-311++G**基组得到了BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH]的垂直电子吸收光谱.所有计算均采用Gaussian 03程序47完成.并采用AIM2000程序48对本文所研究化合物的电子密度进行了拓扑分析.
3 结果与讨论
3.1 BX[(CH2)n]3和 BX(CH2)[CH(CH2)nCH]的结构和稳定性
BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)的对称性、电子态和最小振动频率列于表1中.从表1可以看出,A4、A6和B6三种分子为C1点群对称性,A1、A2和B1的对称性为C3v点群,其它化合物均为C3点群结构,而且这些化合物的电子态均为闭壳层单态.12种化合物的最小振动频率均为正值,表明BX[(CH2)n]3(X=N,P;n=1-6)位于势能面的极小点位置.而且BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)正的最小振动频率表明其位于势能面的极低点.

图1 BX[(CH2)n]3(X=N,P;n=1-6)的优化键长(正体)和Wiberg键级(斜体)Fig.1 Optimized bond lengths(in plain)and Wiberg bond indices(in italic)of BX[(CH2)n]3(X=N,P;n=1-6)bond length in nm
图1为[n.n.n]螺桨烷型分子BX[(CH2)n]3(X=N,P;n=1-6)的几何结构和Wiberg键级.由图1得知,A1中“翻转”的N―B键长为0.1624 nm,Wiberg键级为0.52.A2中桥头B―N键长为0.2401 nm,Wiberg键级仅仅为0.02,这充分说明桥头原子之间不会形成B―N键.而在A3-A5中,中心B―N键长分别为0.1761、0.1703和0.1828 nm,对应的Wiberg键级分别为0.44、0.46和0.43,所以存在B―N键.A6中B―N的键长为0.3815 nm,相应键级为0.02,表明桥头原子之间不存在化学键.相对于B3-B6分子,分子B1的B―P键比较特殊,因为B1中B―P键长比B3-B6的相应键长短,而B1中B―P键级却比B3-B6分子中B―P的键级小.B2中B―P键长为0.2727 nm,比B1中相应键长大0.0786 nm,相应的键级为0.02,因此桥头B和P原子之间没有形成化学键.但在B3-B6中,B―P键长随着桥链的增长从0.1943 nm增大到0.2190 nm,相应的键级分别为0.88、0.90、0.82和0.75.由此可以看出,桥链的增长对中心B―N和B―P键长有影响.其中BP[(CH2)n]3与BN[(CH2)n]3(n=1-6)不同的是BP[(CH2)6]3中形成了B―P键,但BN[(CH2)6]3中不存在B―N键.
图2为BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)[n.n.n]螺桨烷型分子的几何结构和Wiberg键级.从图2可看出,将最小[1.1.1]螺桨烷型分子A1中两个桥链C原子用亚甲基连接得到了它的衍生物C n(n=1-6).C1中B―N键长和键级与A1相比并未发生大的变化.随着亚甲基数目的增多,与C1相比,化合物C2-C6中B―N键长略微减小,键级没有变化.而D1中B―P键长比B1中相应的略有增大,键级基本没有变化.随着亚甲基数目的增多,B―P键长略微减小,键级均为0.38,这和B1中B―P键级基本相同.由此可得知,连接桥链C原子的亚甲基数目对中心B―N和B―P的键长和键级的影响并不大.表1中所示C1-C6和D1-D6的二面角随着亚甲基数目的增多而增大,分别在 95.97°-124.70°和 92.84°-125.05°之间变化.

图2 BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)的优化键长(正体)和Wiberg键级(斜体)Fig.2 Optimized bond lengths(in plain)and Wiberg bond indices(in italic)of BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)bond length in nm

表1 BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)的对称性、电子态、能量(E)、最小振动频率(v min)及BX(CH2)[CH(CH2)nCH]中(HC)-XB-(CH)的二面角Table 1 Symmetries,electronic state,total energy(E),the smallest vibrational frequencies(v min)of BX[(CH2)n]3and BX(CH2)[CH(CH2)nCH],the dihedral angle of(HC)-XB-(CH)in BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)
为了研究BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)的相对稳定性,我们引入化合物平均每增加一个―CH2―时的能量变化和能量的二阶差分.49相对于A1和B1,A n和B n(n=2-6)平均增加一个―CH2―时的能量变化定义为ΔEn=(E1-En)/(3n),而C n和D n(n=2-6)平均增加一个―CH2―时的能量变化定义为ΔEn=(E1-En)/n,E1代表 A1、B1、C1和D1的能量,En代表A n、B n、C n和D n的能量.能量的二阶差分通常用来描述分子或团簇的相对稳定性,能量的二阶差分为Δ2En=En+1+En-1-2En,其中En+1代表 A(n+1)、B(n+1)、C(n+1)和 D(n+1)的能量,而En-1为A(n-1)、B(n-1)、C(n-1)和D(n-1)的能量.

图3 BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P)增加一个CH2时的平均能量Fig.3 Average energy of adding one CH2of BX[(CH2)n]3and BX(CH2)[CH(CH2)nCH](X=N,P)
图3给出了BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P;n=2-6)的ΔEn与n的关系.从图3可看出,所有化合物的ΔEn均随着n值的增大而增大,并且A n、B n、C n和D n的ΔEn的变化是一致的,表明增加CH2的个数到6时有利于稳定这些分子.图4给出了BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH]能量的二阶差分与n的关系.从图4可知,BN[(CH2)n]3能量的二阶差分值在n=3时达到了最大值,BP[(CH2)n]3能量的二阶差分值在n=4时对应的值最大,而BX(CH2)[CH(CH2)nCH](X=N,P)能量的二阶差分在n=2时的值最大,所以A3、B4、C2和D2相对于其它的分子更为稳定.
3.2 BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH]的化学键
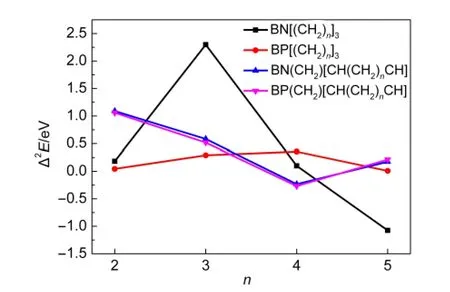
图4 BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P)能量的二阶差分Fig.4 The second-order differences of energies of BX[(CH2)n]3and BX(CH2)[CH(CH2)nCH](X=N,P)

表2 BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)中X和B原子的电子组态以及X(Xe)和B(Be)原子上的电荷分布(e)Table 2 Electron configuration(EC)of X and B,charges(e)on X(Xe),B(Be)of BX[(CH2)n]3and BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)
BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)螺桨烷型分子中X和B的电子组态、电荷分布如表2所示.从表2可以看出,A1中桥头N原子的电子组态为[core]2s1.442p3.97,而A2-A6的接近[core]2s1.302p4.20.A1-A6中N原子的电荷分布在-0.44e至-0.60e之间,B原子上的电荷在0.44e至1.04e之间变化,且由于B―N键长均大于BH3NH3中B―N键长,所以A3-A5中B―N具有离子键特征.B1-B3中P原子组态接近于[core]2s1.602p2.60,而其它三种的均接近于[core]2s1.202p2.50,这和A1-A6中N原子的组态相差比较大.B1-B6中P和B原子的电荷分布范围分别为0.77e至1.29e和0.23e至1.02e,因为B2中B―P的Wiberg键指数只有0.02,所以只有B1、B3-B6中存在P―B键,且具有共价键特征.从表2也可知,C1-C6中桥头N原子的组态接近于[core]2s1.452p3.98,N原子的电荷分布范围为-0.45e至-0.46e,B原子的电荷在0.45e至0.47e之间变化,由此可知C1-C6中均存在具有离子键特征的B―N键.而D1-D6中P原子的组态接近于[core]2s1.602p2.60,桥头P和B原子的电荷分布分别在0.75e至0.77e和0.39e至0.40e之间,所以D1-D6中P―B的键具有共价键特征.
在B3LYP水平上,采用AIM拓扑分析方法计算得到的键临界点(BCP)处电子密度的拓扑分析数据如表3所示,其中包含桥头原子B和N/P之间键鞍点处的电子密度(ρ)和相应的Laplace算符(▽2ρ).从表3可以看出,对于A1、A2、B1和B2四种化合物,AIM计算分析并未找到中心B―N的键临界点和键径.A3、A4和A5中ρ分别为0.092、0.100和0.081,相应的▽2ρ在0.168至0.287范围内变化,这说明桥头B―N具有离子键特征.A6中ρ和▽2ρ分别为0.002和0.007,且B―N键级仅仅为0.02,因此桥头B和N原子之间并不形成化学键.B3-B6四种化合物中ρ在0.122至0.072之间逐渐变化,▽2ρ在-0.098至-0.052范围内变化,所以P―B具有共价键特征,这和NBO分析结果一致.螺桨烷型分子BX[(CH2)n]3(X=N,P;n=1-6)中桥头原子之间的化学键类型和螺桨烷C5H6中C―C键15的明显不同.采用AIM拓扑分析方法,我们没有找到BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)桥头B和N/P之间的键临界点,因而这里没做讨论.

表3 BX[(CH2)n]3(X=N,P;n=1-6)中部分键的电子密度(ρ)和Laplacian值(▽2ρ)Table 3 Density(ρ)and Laplacian(▽2ρ)of selected bonds of BX[(CH2)n]3(X=N,P;n=1-6)
3.3 BX[(CH2)n]3和 BX(CH2)[CH(CH2)nCH]的能隙和电子光谱
图5给出BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)螺桨烷型分子的HOMO-LUMO能隙随n的变化曲线.由图5可知,A1-A6和B1-B6的HOMO-LUMO能隙的变化范围分别在5.24-7.07 eV和5.47-7.33 eV之间,其中A1和B1的能隙最大(>7.0 eV),与C5H6的能隙7.27 eV很接近,而A2和B2的能隙最小(在5.20-5.50 eV之间).化合物C1-C6和D1-D6的HOMO-LUMO能隙分别在6.90和6.70 eV附近,且C n的HOMO-LUMO能隙均高于D n的能隙.

图5 BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)的能隙Fig.5 Energy gaps of BX[(CH2)n]3and BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)

图6 BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)的第一垂直激发能Fig.6 The first vertical transition energies of BX[(CH2)n]3and BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)
BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)螺桨烷型分子的垂直电子跃迁波长随n的变化曲线列于图6中.从图6可知,随着n的变化,A n和B n的激发能变化很大,而C n和D n的激发能变化很小.A1-A6的激发能位于191.1-284.8 nm之间.B1-B6的垂直跃迁能位于191.8-270.1 nm之间,C1-C6和D1-D6的垂直跃迁能分别在190.5-199.7 nm和209.0-221.3 nm之间.
4 结论
采用密度泛函理论得到了螺桨烷型分子BX[(CH2)n]3和BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)的稳定结构和电子光谱,并采用NBO和AIM方法探讨了它们的化学键性质.所研究体系均位于势能面的极小点,其能隙均大于5.20 eV,特别是,BN[CH2]3和BP[CH2]3的能隙与C5H6的能隙很接近.而且从分子能量的二阶差分得到BN[(CH2)3]3、BP[(CH2)4]3以及 BX(CH2)[CH(CH2)2CH](X=N,P)是最稳定的.BN[(CH2)n]3中只有n=1,3,4,5时存在B―N键,BP[(CH2)n]3中只有n=1,3,4,5,6时存在B―P键,但是BX(CH2)[CH(CH2)nCH](X=N,P;n=1-6)中桥头两原子均存在化学键.BN[(CH2)n]3、BP[(CH2)n]3、BN(CH2)[CH(CH2)nCH]和BP(CH2)[CH(CH2)nCH](n=1-6)的垂直激发能的范围分别为191.1-284.8 nm、191.8-270.1 nm、190.5-199.7 nm和209.0-221.3 nm.
(2)Wiberg,K.B.;Walker,F.H.J.Am.Chem.Soc.1982,104,5239.doi:10.1021/ja00383a046
(3)Wiberg,K.B.;Walker,S.T.;Rosenberg,R.E.J.Am.Chem.Soc.1990,112,2184.doi:10.1021/ja00162a021
(4)Jackson,J.E.;Allen,L.C.J.Am.Chem.Soc.1984,106,591.doi:10.1021/ja00315a022
(5)Feller,D.;Davidson,E.R.J.Am.Chem.Soc.1987,109,4133.doi:10.1021/ja00248a001
(6)Mcgarry,P.F.;Johnsoton,L.J.;Scaiano,J.C.J.Org.Chem.1989,54,6133.doi:10.1021/jo00287a033
(7)Wiberg,K.B.Accounts Chem.Res.1984,17,379.doi:10.1021/ar00107a001
(8)Wiberg,K.B.J.Am.Chem.Soc.1983,105,1227.doi:10.1021/ja00343a025
(9)Epiotis,N.D.J.Am.Chem.Soc.1984,106,3170.doi:10.1021/ja00323a018
(10)Zhao,C.Y.;Wei,T.S.;Qiu,W.Y.Acta Chim.Sin.1991,49,546.[赵存元,韦统帅,邱文元.化学学报,1991,49,546.]
(11)Honegger,E.;Huber,H.;Heilbronner,E.;Dailey,W.P.;Wiberg,K.B.J.Am.Chem.Soc.1985,107,7172.doi:10.1021/ja00310a068
(12)Messmer,R.P.;Schultz,P.A.J.Am.Chem.Soc.1986,108,7407.doi:10.1021/ja00283a045
(13)Riggs,N.V.;Zoller,U.;Nguyen,M.T.;Radom,L.J.Am.Chem.Soc.1992,114,4354.doi:10.1021/ja00037a048
(14)Wu,W.;Gu,J.;Song,J.;Shaik,S.;Hiberty,P.C.Angew.Chem.Int.Edit.2009,48,1407.doi:10.1002/anie.v48:8
(15)Danovich,S.D.;Wu,W.;Hiberty,P.C.Nat.Chem.2009,1,443.doi:10.1038/nchem.327
(16)Shaik,S.;Chen,Z.H.;Wu,W.;Stanger,A.;Danovich,D.;Hiberty,P.C.ChemPhysChem 2009,10,2658.doi:10.1002/cphc.v10:15
(17)Gershoni-Poranne,R.;Stanger,A.ChemPhysChem 2012,13,2377.doi:10.1002/cphc.v13.9
(18)Eaton,P.E.;Temme,G.H.J.Am.Chem.Soc.1973,95,7508.doi:10.1021/ja00803a052
(19)Wiberg,K.B.;Walker,F.H.;Michl,J.J.Am.Chem.Soc.1982,104,2056.doi:10.1021/ja00371a059
(20)Hamon,D.P.G.;Trenerry,V.C.J.Am.Chem.Soc.1981,103,4962.doi:10.1021/ja00406a059
(21)Szeimies,S.U.;Szeimies,G.J.Am.Chem.Soc.1978,100,3966.doi:10.1021/ja00480a072
(22)Gassman,P.G.;Proehl,G.S.J.Am.Chem.Soc.1980,102,6862.doi:10.1021/ja00542a040
(23)Mlinaric-Majerski,K.;Majerski,Z.J.Am.Chem.Soc.1980,102,1418.doi:10.1021/ja00524a033
(24)Wiberg,K.B.;Burgmaier,G.J.Am.Chem.Soc.1972,94,7396.doi:10.1021/ja00776a022
(25)Aue,D.H.;Reynolds,R.N.J.Org.Chem.1974,39,2315.doi:10.1021/jo00929a051
(26)Schleyer,P.v.R.;Janoschek,R.P.Angew.Chem.1987,26,1267.
(27)Nagase,S.;Kudo,T.Organometallics 1987,6,2456.doi:10.1021/om00154a034
(28)Tsumuraya,T.;Batcheller,S.A.;Masamune,S.Angew.Chem.1991,30,902.
(29)Gallego-Planas,N.;Whitehead,M.A.J.Mol.Struct.-Theochem 1992,260,419.doi:10.1016/0166-1280(92)87057-7
(30)Holder,A.J.;Earley,C.W.J.Mol.Struct.-Theochem 1993,281,131.doi:10.1016/0166-1280(93)87070-T
(31)Karni,M.;Apeloig,Y.;Kapp,J.;Schleyer,P.v.R.In The Chemistry of Organic Silicon Compounds;John Wiley&Sons:New York,2001;pp 1-163.
(32)Nied,D.;Koppe,R;.Klopper,W.;Schnockel,H.;Breher,F.S.J.Am.Chem.Soc.2010,132,10264.doi:10.1021/ja104810u
(33)Nied,D.;Oña-Burgos,P.;Klopper,W.;Breher,F.Organometallics 2011,30,1419.doi:10.1021/om100977z
(34)Richards,A.F.;Brynda,M.;Power,P.P.Organometallics 2004,23,4009.doi:10.1021/om0497137
(35)Ito,Y.;Lee,V.Y.;Gornitzka,H.;Goedecke,C.;Frenking,G.;Sekiguchi,A.J.Am.Chem.Soc.2013,135,6770.doi:10.1021/ja401650q
(36)Yoshida,H.;Takahara,Y.;Erata,T.;Ando,W.J.Am.Chem.Soc.1992,114,1098.doi:10.1021/ja00029a055
(37)Monakhov,K.Y.;Gourlaouen,C.Organometallics 2012,31,4415.doi:10.1021/om300180e
(38)Daly,A.M.;Tanjaroon,C.;Marwitz,A.V.;Liu,S.Y.;Kukolich,S.G.J.Am.Chem.Soc.2010,132,5501.
(39)Liu,Z.Q.;Marder,T.B.Angew.Chem.Int.Edit.2008,47,242.
(40)Song,Y.;Chen,H.S.;Zhang,C.R.;Wang,G.H.Acta Phys.-Chim.Sin.2005,21,735.[宋 燕,陈宏善,张材荣,王广厚.物理化学学报,2005,21,735.]doi:10.3866/PKU.WHXB20050708
(41)Hohenberg,P.;Kohn,W.Phys.Rev.1964,136,B864.
(42)Kohn,W.;Sham,L.Phys.Rev.1965,140,A1133.
(43)Becke,A.D.J.Chem.Phys.1993,98,5648.doi:10.1063/1.464913
(44)Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B 1988,37,785.doi:10.1103/PhysRevB.37.785
(45)Reed,A.E.;Curtiss,L.A.;Weinhold,F.Chem.Rev.1988,88,899.doi:10.1021/cr00088a005
(46)Casida,M.E.;Jamorski,C.;Casida,K.C.;Salahub,D.R.J.Chem.Phys.1998,108,4439.doi:10.1063/1.475855
(47)Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et.al.Gaussian 03,Revision C.02;Gaussian Inc.:Wallingford,CT,2003.
(48)Biegler-Koning,F.J.;Derdau,R.;Bayles,D.;Bader,R.F.W.AIM2000,Version1,2000.
(49)Lei,X.L.;Zhu,H.J.;Wang,X.M.;Luo,Y.H.Acta Phys.-Chim.Sin.2008,24,1655.[雷雪玲,祝恒江,王先明,罗有华.物理化学学报,2008,24,1655.]doi:10.3866/PKU.WHXB20080922
猜你喜欢
物理学报(2021年12期)2021-07-01
数学物理学报(2020年6期)2021-01-14
青岛大学学报(工程技术版)(2019年2期)2019-09-10
考试周刊(2016年60期)2016-08-23
科技资讯(2016年5期)2016-08-13
考试周刊(2016年48期)2016-06-29
中学生数理化·高二版(2016年6期)2016-05-14
武汉工程大学学报(2016年1期)2016-04-07
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
