
黑龙江省和吉林省稻瘟病菌种群多样性研究
2014-10-16 08:53张亚玲王宝玉台莲梅左豫虎郑雯邓本良靳学慧
黑龙江八一农垦大学学报 2014年1期
张亚玲,王宝玉,台莲梅,左豫虎,郑雯,邓本良,靳学慧
(黑龙江八一农垦大学农学院,大庆163319)
稻瘟病是由稻瘟病菌Magnaporthe oryzae B.Couch(无性态:Pyricularia oryzae Cavara以前为Magnaporthe grisea(Hebert))Barr.引起的,是对水稻产量损失影响最大的一种病害[1-2]。长期实践表明,控制此病害最经济环保的措施是培育和合理利用抗病品种[3],然而由于稻瘟病菌群体发生变异而引发新的致病小种的出现导致了新选育品种的抗性只能维持短期的效应[4]。因此研究稻瘟病菌群体结构能够更好地了解稻瘟病菌在田间的组成,对于抗病品种的选育和合理使用有一定的理论意义。对于稻瘟病菌种群结构研究人们利用最多的方法是致病性鉴定的方法进行,对稻瘟病菌的群体结构研究起到了重要的作用,但随着生物技术的发展生物技术手段已经完全渗入到生物类群体结构研究中。Hamer等[5]从稻瘟病菌中分离得到一组散布中等重复序列的DNA片段,在稻瘟菌基因组中有丰富的多态性。从此,DNA指纹分析在稻瘟病菌的群体结构分析中得到广泛应用。Pot2-rep-PCR是基于中等重复序列的一种PCR扩增技术[6]。利用Pot2-rep-PCR技术对我省水稻主产区的49个稻瘟病菌菌株进行群体遗传分析,为北方稻区水稻抗瘟品种的合理使用奠定理论基础。
1 材料与方法
1.1 供试菌株
参试的菌株共有49个,其中黑龙江省稻菌株23个,吉林稻瘟病菌菌株26个(表1)。

表1 试验菌株Table1 Magnaporthe grisea Isolates used in the study and serial number
1.2 病原菌的单孢分离
取稻瘟病穗颈瘟发病部位于培养皿内保湿培养24 h,当发病部位产生孢子后在PDA(1 000 mL水,15 g葡萄糖,12 g琼脂粉)培养基上采用震落的方法使孢子落于平板培养基上,于25℃下培养36 h,长出菌落后在无菌条件下通过显微镜调取单菌落。
1.3 菌丝的培养及DNA的制备
将供试菌株活化于米糠培养基上,培养10 d后,挑取4块新鲜菌丝块(0.5×0.5 cm)接种于经高压灭菌的液体酵母培养基中。然后置于摇床中28℃恒温,170 rpm振荡培养2~3 d,待菌丝体生长茂盛且未变黑时,用灭菌的纱布和滤纸真空过滤,再用灭菌蒸馏水抽洗2次,最后用滤纸尽可能地将菌丝中的水吸干,并分装于1.5mL离心管中,置于-20℃冰箱中冷冻保存。
将研钵洗净,于120℃干热灭菌,冷却备用。取约150 mg的冻干菌丝置于预冷的研钵中,加入液氮,研磨(3~4次)至粉末状,然后,用DNA抽提试剂盒(E.Z.N.A.〇RFungal DNA Kit,Omega公司,美国)对基因组DNA进行抽提,其提取方法参照试剂合说明书步骤进行。
1.4 Pot2-Rep-PCR扩增
扩增引物序列:Pot2-1 5’-CGGAAGCCCTAAA GCTGTTT-3’Pot2-2 5’-CCCTCATTCGTCAC ACGTTC-3’
由Sangon(上海生工生物工程技术服务有限公司)公司合成。扩增体系及反应过程如下:
扩增所采用25μL体系。灭菌双纯水12.5μL,引物Pot2-1(25 pmol),Pot2-2(25 pmol)各1μL,dNTP(mM)1μL,含水量Mg2+Buffer 2μL,模板1μL,DNA Taq 0.2μL。反应循环:94℃5min,变性95℃2.5 min;4个循环的变性94℃1 min,退火62℃1min,延伸65℃10min;26个循环的变性94℃30 s,退火62℃1min,延伸65℃10min;最后65℃延伸15min。扩增完毕后取12.5μL在1.5%琼脂糖凝胶上电泳。
1.5 扩增结果检测
rep-PCR扩增产物用1.5%琼脂糖凝胶电泳检测,Bio-rad凝胶成像系统照相。
1.6 数据处理
把电泳照片上的扩增条带转换成2进制数据,即有条带记为“1”,无条带记为“0”;按Percent disagreement遗传距离公式计算相似系数;然后用Statistica中的UPGA(Unweightedpair-group average)聚类分析得到反映菌株亲缘关系的树状图。
2 结果与分析
2.1 rep-PCR遗传谱型分析
用一对引物(Pot2-1/Pot2-2)对49个供试菌株均有较好扩增效果,扩增产物经电泳检测,可获得稳定、清晰明亮的谱带。结果显示所有供试菌株分别扩增到1~16条带,大小从100 bp到3 kb之间(图1),特异性谱带共有16条,没有发现49个菌株都共有的保守带,可见稻瘟病菌群体存在丰富的遗传多样性。说明稻瘟菌在DNA水平上具有高度的异质性。利用UPGA聚类分析,所有的菌株遗传距离在0.20水平上可将供试的菌株划分为20个遗传谱,其中第2个谱系为优势谱系,黑龙江省有11个菌株,吉林省有9个菌株;其次是谱系1有黑龙江省1个菌株,吉林3个菌株;其中谱系2是绝对优势系谱,含有的单型数目超过40%;其他19个系谱的单型数都为1~10。
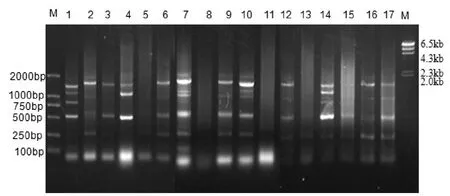
图1 部分稻瘟病菌菌株PCR电泳图Fig.1 Partof the rice blast fungus strains PCR electrophoregram
2.2 不同稻区稻瘟病菌遗传多样性
分析结果表明,20个系谱内含有的菌株数差异明显,第2个谱系是优势谱系,黑龙江省的吉林省的供试菌株以接近于1∶1的组成分布在这个谱系内;第1谱系内有4个菌株,其中有3个是吉林省菌株。从图2可以看出吉林菌株JMTY130-1菌株与其他菌株明显不同,在扩增时能扩增出条带,只能扩出2条,其他菌株扩增4~12条不等。虽然供试菌株数量不多,但在一定程度上显现出菌株遗传背景不同。指纹图谱遗传相似性聚类分析显示:地区间差异极其明显。以0.20相似性水平划分遗传系谱,49个菌株可划分在20个系谱中(图2)。根据聚类分析黑龙江省和吉林省参试菌株所占系谱数与参试菌株数的比例来看,参试稻瘟菌株遗传多样性程度黑龙江省(系谱数/菌株数)黑龙江省(7/23=0.304 3)、吉林省(11/26=0.423 0),这个研究结果虽然与采样数量有关,但也间接反应了黑龙江与吉林两省稻瘟病菌的遗传多样性的程度,从两个省的菌株在谱系中有交叉存在的现象也说明黑龙江省菌株和吉林菌的遗传比较相似。谱系3主要以黑龙江省菌株存在,说明谱系3是黑龙江省特有的谱系类型。

图2 49个供试稻瘟病菌菌株遗传距离聚类图Fig.2 Tree diagram derived from gengetic of 49 strains of Pyricularia oryzae Cav
3 讨论
人们采用多种方法对稻瘟病菌遗传多样性进行研究,如SRAP方法[3]、RAPD方法[4]、RFLP方法[5-6]等,随着生物技术的发展,研究人员研究发现rep-PCR技术是简单实用的一种研究稻瘟病菌种群多样性的技术[7-8],周益军等[9]利用Pot2-rep-PCR技术对收集自我国14个省水稻产区的324个稻瘟病菌株进行了DNA指纹分析。稻瘟病菌在DNA水平上的变异比较高,具有很丰富的多态性,在20%的遗传距离水平上,170个单型被划分成20个遗传系谱。2004年周益军等[10]对亚洲五国的稻瘟病菌遗传多样性进行了研究,将不同地点的稻瘟病菌划分为不同的谱系。这些研究结果都说明利用Pot2-rep-PCR技术对稻瘟病菌群体组成的研究有应用价值。
稻瘟病菌种群结构差异的原因我多种可能,但主要分为两大类,一类是气候条件,另一类是品种及品种的合理使用[11]。作者认为稻瘟病菌的变异受多种因素共同影响,是一个长期进化过程,稻瘟病菌群体结构因年度和地区变化是很显著的,稻瘟病菌具有高度的遗传多样性和变异性及较强的时空局限性[6]。通过对黑龙江省和吉林省稻瘟病菌种群结构研究结果表明,两省稻瘟病菌的种群结构存在丰富的多样性,吉林省遗传多样性要强一些,这可能与吉林省地理位置在黑龙江省以南,受气候条件影响,吉林省可种植的品种要比黑龙江多些,这对稻瘟病菌的种群结构也会产生影响。另外研究所采用的菌株数量有限,不一定能完全说明黑龙江省和吉林省稻瘟病菌遗传多样性差异,可能会与田间实际情况有所不同,为进一步分析稻瘟病菌在田间的种群多样性还需要大量采集代表地区稻瘟病菌做进一步的研究分析。
Pot2-rep-PCR是基于中等重复序列的一种PCR扩增技术[12],具有简单,分辨能力高的优点,在DNA指纹分析在稻瘟病菌的群体结构分析中得到广泛应用,有许多的研究者利用Pot2-rep-PCR对国内外各不同稻区稻瘟菌株进行了分析,得到了不同的研究结果,由于各自的研究分析方法有所不同,所得结果不能进行比较分析。因此,探索发现其他可能途径以对病原菌DNA指纹进行分析是目前病原菌群体结构研究的重点。
[1]Couch B C,Kohn L M A.Multilocus gene genealogy concordant with host preference indicates segregation of a new species,Magnaporthe oryzea from M.grisea[J].Mycologia,2002,94(4):683-693.
[2]Ou S H.Rice diseases[M].2th ed.UK:Commonwealth Mycological Institute,1985.
[3]张俊华,刘扬,孙供利,等.黑龙江省稻瘟病菌遗传多样性研究[J].东北农业大学学报,2013,44(1):34-38.
[4]吕军.黑龙江省稻瘟病菌致病性分化与RAPD分析[D].大庆:黑龙江八一农垦大学,2007.
[5]De Bruijn F J.Use of repetitive(repetitive extragenic palindromic and enterobacterial repetitive intergeneric consensus)sequences and the polymerase chain reaction to fingerprint the genomes of Rhizobium meliloti isolates and other soil bacteria[J].Appl.Environ.Microbiol,1992,58(7),2180-2187.
[6]George M L C,Bustamam M,CruzW T,et al.Movement of Xanthomonas oryzae pv.oryzae in southeast Asia detected using PCR-based DNA fingerprinting[J].Phytopathology,1997,87(3):302-309.
[7]Levy M,Romao J,Marchetti M A,et al.DNA fingerprinting with a dispersed repeated sequence resolves pathotype diversity in the rice blast fungus[J].PlantCell,1991,3(1):95-102.
[8]周益军,白娟,程兆榜,等.我国稻瘟病菌群体多样性研究[J].中国水稻科学,2004,18(3):277-280.
[9]周益军,陈葆棠,程兆榜.亚洲5国稻瘟病菌(Magnaporthe grisea)遗传多样性初探[J].中国水稻科学,2004,20(3):194-196.
[10]刘二明.水稻品种与稻瘟病菌的遗传多样性及稻瘟病持续控制研究[D].成都:四川农业大学,2001.
[11]George M L C,Nelson R J,Zeigler R S,et al.Rapid population analysis of Magnaporthe grisea by using rep-PCR and endogenous repetitive DNA sequences[J].Phytopathology,1998,88(3):223-229.
猜你喜欢
艺术品鉴(2022年16期)2022-07-09
作物学报(2022年6期)2022-04-08
华北电力大学学报(社会科学版)(2021年2期)2021-07-21
河池学院学报(2021年1期)2021-07-10
景德镇陶瓷(2021年1期)2021-03-24
图书馆理论与实践(2018年2期)2018-01-28
东方考古(2017年0期)2017-07-11
中国奶牛(2017年2期)2017-03-22
求学·理科版(2017年1期)2017-03-02
河南农业(2016年6期)2016-11-26
