
红花药材中非法染有酸性红73的TLC鉴别和HPLC确证
2014-09-26 07:45杨荣艳宋瑩李本淳
中国现代中药 2014年8期
杨荣艳,宋瑩,李本淳
(吉林省四平市食品药品检验所,吉林 四平 136000)
红花药材中非法染有酸性红73的TLC鉴别和HPLC确证
杨荣艳*,宋瑩,李本淳
(吉林省四平市食品药品检验所,吉林 四平 136000)
目的:建立红花药材中非法染有酸性红73的检测方法。方法:采用TLC对红花中非法染有酸性红73进行定性鉴别;采用HPLC确证建立的TLC可行。结果:在TLC中,阳性样品可检出与酸性红73对照试剂位置和颜色一致的斑点;HPLC中,也出现了与对照试剂保留时间一致的色谱峰,并且DAD检测器检验,在420~580 nm波长范围的紫外-可见吸收光谱相同。结论:该方法前处理简便、快速、准确,可用于红花药材中非法染有酸性红73的检查。
红花;酸性红73;薄层色谱法;高效液相色谱法
红花收载于《中国药典》2010年版一部,检验项目有性状、显微鉴别、薄层鉴别、水分检查、杂质检查、总灰分检查、酸不溶性灰分检查、吸光度检查、浸出物和含量测定[1]。《药品检验补充检验方法和检验项目批准件(编号2007009)》[2]中补充了红花金橙Ⅱ检查项。在2012年度药品抽验中,笔者按照《中国药典》2010年版一部对20批红花进行薄层鉴别检验时,发现有3批红花供试品色谱比对照药材色谱相应的位置上多一个红色斑点,并对此斑点的成分进行了研究,初步确认为酸性红73。酸性红73是一种强烈的致癌物质,被国家禁止用于食品中的工业染料,威胁患者的健康[3]。笔者通过实验建立薄层色谱定性鉴别和高效液相色谱确证红花中非法染有酸性红73的方法,为药品监管提供技术支持。
1 仪器与试药
1.1 仪器
LC-2010CHT型高效液相色谱仪(岛津),紫外检测器和DAD检测器,CLASS-VP色谱工作站,UV-2450可见-紫外分光光度计,BP211D型电子分析天平,LC-250超声清洗器(功率250 W,频率50 kHZ);纯水器(德国)。
1.2 试药
酸性红73对照试剂(美国AccuStandard公司,供含量测定用,批号:16675);色谱甲醇,自制超纯水,醋酸铵、乙醇均为分析纯,市售硅胶G板(上海盛亚化工有限公司,批号:20090306)。
红花(市售),红花对照药材(中国食品药品检定研究院,批号:120907-201111)。
2 方法与结果
2.1 薄层色谱鉴别[4]
取本品1 g,加70%乙醇20 mL,超声提取20 min,取上清液作为供试品溶液。另取酸性红73对照试剂适量,分别加70%乙醇溶解并制成每1 mL含60 μg的溶液,作为对照试剂溶液。照《中国药典》2010年版一部薄层色谱法(附录Ⅵ B)试验,吸取供试品溶液和对照试剂溶液各5 μL,分别点于同一硅胶G薄层板上,以乙酸乙酯-正丁醇-乙醇-氨水-水(1∶3∶3∶1∶1)为展开剂,展开,取出,晾干,在可见光下检视。供试品色谱中,在与对照试剂色谱相应的位置上,不显相同颜色的斑点;阴性对照不干扰测定。见图1。

1~3.供试品 4.酸性红73对照试剂 5.红花对照药材图1 红花及对照品TLC图
2.2 HPLC测定酸性红73(紫外检测器)
2.2.1 色谱条件[5-6]色谱柱:Agilent TC-C18(250 mm×4.6 mm,5 μm),流动相:甲醇-0.025 mol·L-1醋酸铵溶液,甲醇梯度洗脱条件:20%~45%(0~8 min),45%~80%(8~20 min);流速:1.0 mL·min-1,检测波长:508 nm,进样量:10 μL,柱温:35 ℃。2.2.2 对照品溶液的制备 酸性红73对照试剂12 mg,精密称定,置200 mL量瓶中,加70%乙醇稀释至刻度,即得(每1 mL含酸性红73对照试剂60 μg)。
2.2.3 供试品溶液的制备[5,7]取本品约1 g,加70%乙醇20 mL,超声处理20 min,放冷,取续滤液,即得。
2.2.4 阴性样品溶液的制备 取不含有酸性红73的红花对照药材作为阴性样品,按照2.2.3供试品溶液的制备方法制备阴性样品溶液。
2.2.5 专属性试验 把供试品溶液、阴性样品溶液和酸性红73对照试剂分别注入高效液相色谱仪,依法测定,结果供试品色谱图中,供试品主峰保留时间与酸性红73对照试剂主峰保留时间一致;阴性样品色谱图中,在与酸性红73对照试剂主峰保留时间处无峰出现。见图2。

A.酸性红73对照试剂 B.供试品 C.红花对照药材图2 红花及对照品HPLC图
2.2.6 线性关系的考察 精密称取酸性红73对照试剂(含量测定用)10.10 mg,置100 mL容量瓶中,加用0.45 μm的微孔滤膜过滤后的70%乙醇适量,超声溶解并定容至刻度,摇匀,作为对照试剂溶液储备液。
精密量取对照试剂溶液储备液0.1,0.5,1,2,3,6,8,10 mL分别置10 mL量瓶中,用上述70%乙醇定容至刻度,摇匀,即得质量浓度分别为1.01,5.05,10.10,20.20,30.30,60.60,80.80,100.10 μg·mL-1系列标准溶液。不经微孔滤膜过滤,分别将上述系列标准溶液各10 μL注入高效液相色谱仪分析测定,每个浓度连续进样3次,取3次平均值,以质量浓度为横坐标(X),峰面积为纵坐标(Y),经线性回归,得回归方程Y=30 393X-2 583.8,r=1.000。结果表明,酸性红73在1.01~100.10 μg·mL-1具有良好的线性关系。
2.2.7 精密度试验 取同一质量浓度(60 μg·mL-1)对照品溶液按上述色谱条件测定,连续进样6次,结果峰面积RSD=0.34%(n=6),表明仪器精密度良好。
2.2.8 稳定性试验 取同一供试品溶液,照上述色谱条件测定,考察了0,2,4,8,12,16,24 h峰面积值的变化,结果峰面积RSD=1.18%(n=6),表明样品溶液在24 h内稳定。
2.2.9 重复性试验 取同一供试品12份,共分为2组,每份1.0 g,精密称定,精密加入70%乙醇20 mL,照2.2.3方法制备,独立试验,结果第一组中酸性红73的平均质量分数为0.40 mg·g-1,RSD=5.12% (n=6);第二组中酸性红73的平均质量分数为0.48 mg·g-1,RSD=8.03%(n=6)。
2.2.10 阴性样品回收率试验 称取红花对照药材样品6份,每份约0.5 g,精密称定,分别置具塞锥形瓶中,分别精密加入对照试剂溶液10 mL(60 μg·mL-1),再分别精密加入70%乙醇10 mL,称定重量,超声处理20 min,放冷,补足减失的重量,摇匀,用0.45 μm的微孔滤头(用待过滤液4 mL使滤头充分饱和)过滤,进样量10 μL,注入高效液相色谱仪测定,即得。测定结果见表1。
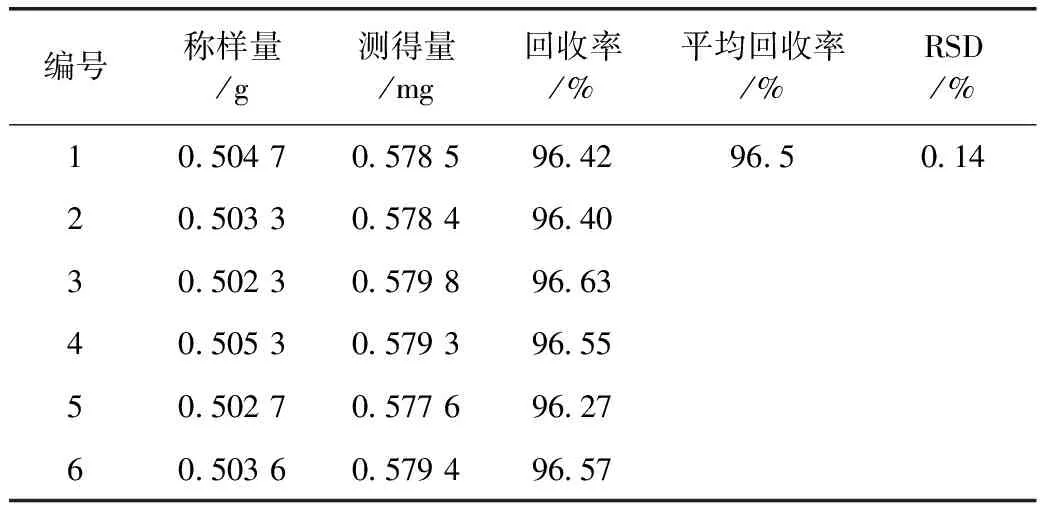
表3 红花阴性样品回收率试验
注:样品中含酸性红73的量均为0 mg,酸性红73对照试剂加入量均为0.600 0 mg
2.2.11 方法的检测限 按照正文色谱条件,基线走稳后,进一针10 μL溶剂,再分别将质量浓度为0.1,0.2,0.3 μg·mL-1的对照试剂各10 μL注入高效液相色谱仪,结果0.3 μg·mL的对照试剂信噪比S/N=3,所以将方法的检测限定为0.3 μg·mL-1。
2.2.12 样品测定 照上述色谱条件和制备方法,依法测定3批市售红花样品,分别测定3次,3批样品中酸性红73的质量分数分别为0.45,0.71,0.86 mg·g-1。
2.3 高效液相色谱法DAD检测器确证
2.3.1 色谱条件、供试品和对照品试剂的制备 DAD检测器,其他条件同2.2.1;供试品和对照品试剂的制备分别同2.2.2和2.2.3。
2.3.2 高效液相色谱法DAD检测器检验 供试品色谱中,出现与对照试剂保留时间一致的色谱峰。采用二极管阵列检测器比较相应色谱峰在420~580 nm波长范围的紫外-可见吸收光谱,发现吸收光谱相同。见图3~6。

图3 酸性红73对照试剂光谱

图4 酸性红73对照试剂色谱

图5 红花供试品光谱

图6 红花供试品色谱
3 讨论
实验中发现两组重复性试验结果RSD值均大于3.0%,为此考察了样品粉碎与否对结果的影响,结果发现不粉碎的样品含量高于粉碎样品的含量,样品处理得越细,含量越低,而且含量也不均匀。考虑到酸性红73是一种着色剂,一般情况下都会附着在样品的表面,所以采取不粉碎处理样品。
另外,实验中发现不同浓度的对照试剂溶液经0.45 μm微孔滤头过滤后,峰面积与浓度比不成正比,考虑到可能是微孔滤头对酸性红73具有吸附作用。我们采用同浓度对照试剂溶液,通过过滤和未过滤来验证是否具有吸附作用,考察结果发现,未经过滤的对照试剂的峰面积是经过滤的对照试剂的峰面积的1.2倍,损失率约占16%。考虑到每次用滤头过滤供试品溶液时,其对供试品溶液中酸性红73吸附程度不同,从而导致供试品含量不确定。上述结果说明样品本身的均匀性差和酸性红73的吸附性是影响重复性试验结果的原因。实验方法只是针对非法染有色素酸性红73的红花样品检查,并非含量测定项,虽然RSD超出3.0%,但并不影响方法的应用。
通过12份样品的重复性试验发现,红花样品中酸性红73的质量分数在0.38~0.52 mg·g-1,表明样品本身的均匀性很差,如采用样品加标,无法得到准确的回收率结果,所以我们采取阴性样品加标进行回收试验,结果RSD=0.14%,符合《中国药典》规定的要求,说明此检验方法可行。
考察了shimpack C18、zorbax SB C18、Agilent TC-C183种色谱柱,结果都能得到很好的色谱分离。另外,考察了流动相中醋酸铵溶液的两种浓度即0.025,0.05 mol·L-1,结果都能达到系统适用性要求,但是考虑到含高浓度盐的流动相对色谱柱伤害较大,所以采用0.025 mol·L-1醋酸铵溶液。
实验建立的检验方法前处理简便、快速、准确,可用于红花药材中非法染有酸性红73的检查,研究为红花监管提供技术支持。
[1] 国家药典委员会.中国药典[S].一部.北京:中国医药科技出版社,2010:141-142.
[2] 国家食品药品监督管理局.药品检验补充检验方法和检验项目批准件汇编(2003~2008年)[S].2007:154.
[3] 国家食品药品监督管理局稽查局.五味子补充检验方法和检验项目批准件[S].2007
[4] 闵春艳,付凌燕,汪祺,等.红花药材掺伪染色检测方法的实验研究[J].中国药事,2011,(8):772-775.
[5] 肖海龙,屠海云,王红青,等.反相高效液相色谱法快速测定食品中18种水溶性合成着色剂[J].中国卫生检验杂志,2011,21(2):264-266.
[6] 邹耀华,殷红妹,郭怡飚,等.HPLC-PDA法检测西红花和红花中十一种非法添加色素[J].中国卫生检验杂志,2010,(11):2724-2725.
[7] 汪建君,陈惠玲.HPLC-PDA法检测正天丸和正天胶囊中6种非法添加色素[J].药物评价研究,2013,(1):51-53.
TLCHPLCIdentificationofillegallystainedAcidRed73inCarthamiFlos
YANG Rongyan*,SONG Ying,LI Benchun
(SipingInstituteforDrugandFoodControl,Siping136000,China)
Objective:To establish TLC identification and HPLC confirmatory method for illegally stained Acid Red 73 in Carthami Flos.Methods:TLC was employed to identify Acid Red 73,which was confirmed with HPLC.Results:In TLC,the emergence of Acid Red 73 with a consistent position and color spots.In HPLC,the emergence of the peak retention time,and comparing the wavelength range of 420-580 nm UV-visible absorption spectrum by a DAD detector,the same absorption spectrum was found.Conclusion:The method is easy,quick and accurate,can be used to identify Acid Red 73 in Carthami Flos.
Carthami Flos;Acid Red 73;TLC;HPLC
10.13313/j.issn.1673-4890.2014.08.005
2013-09-12)
*
杨荣艳,硕士,主管药师,研究方向:中药检验;E-mail:yry_lg@126.com
猜你喜欢
快乐语文(2021年34期)2022-01-18
快乐语文(2021年27期)2021-11-24
现代临床医学(2021年6期)2021-11-20
快乐语文(2021年11期)2021-07-20
快乐语文(2021年15期)2021-06-15
现代仪器与医疗(2021年1期)2021-06-09
保健文汇(2020年8期)2020-12-02
福建基础教育研究(2019年8期)2019-05-28
中国资源综合利用(2017年4期)2018-01-22
中学生数理化·高二版(2016年3期)2016-12-26
