
金属有机骨架材料MIL-53(Al)负载钴催化剂的CO 催化氧化反应性能
2014-09-21 08:59谭海燕吴金平
物理化学学报 2014年4期
谭海燕 吴金平
(中国地质大学(武汉)材料与化学学院,可持续能源实验室,武汉430074)
1 引言
空气污染是当今社会关注的重要议题之一,污染成份中的CO是大气中分布最广和数量最多的污染物之一,1汽车尾气排放物中含有的CO以及碳的不完全燃烧,都加大了空气中CO气体的含量,除去汽车尾气中的CO显得尤为重要.目前对CO的消除通常采用催化氧化法.2CO氧化反应因其相对简单具有代表性,在理论研究中通常被作为催化研究中的探针反应,因此CO的催化氧化反应是兼有理论研究意义和实际应用价值的化学反应.3在CO催化氧化的研究中,贵金属催化剂具有很好的活性和稳定性,4,5但是它们昂贵的价格使得越来越多的研究者开始考虑使用非贵金属催化剂,常见的非贵金属如Cu、Co、Ni等.6在钴的负载型催化剂中,一般载体的比表面积越大,孔容越大,越能将金属更好地分散,在反应中的催化效果越好.7但传统的载体材料如Al2O3、SiO2的比表面积约为200-300 m2·g-1,这些载体的比表面积有待提高.
金属有机骨架材料(MOFs)是近十年来受到学术界广泛关注的一类新型纳米多孔材料,8-10它是有机配体与金属离子通过自组装过程形成的具有周期性网络结构的骨架材料.它的比表面积很大,孔径可调,被广泛用作吸附材料、11催化材料、12-14磁性材料、光学材料和电池材料等.15-17与传统的催化剂载体相比,MOFs的优点是结构可调(通过调变有机物配体来改变孔径大小和结构),比表面积高并且具有较大的孔容.由于它的比表面积高,可以通过浸渍、共沉淀和溶胶-凝胶等方法将金属负载到MOFs的表面或孔道中.MOFs作为一类具有特殊结构的新型催化剂载体已引起了催化研究者的广泛关注,并被用于CO的催化氧化.Zou等18,19首次将MOFs作为催化材料用于CO的催化氧化反应,他们合成的多孔材料[Cu(mipt)(H2O)](H2O)2在200°C时使CO转化率达到100%.Jiang等20用稳定性高、比表面积大的ZIF-8作载体负载金纳米颗粒用于CO催化氧化,在315-340°C时使CO转化率达到100%.最近,Cu3(BTC)2(又叫HKUST)和MIL-101被用作载体,分别负载Ce(NO3)3·6H2O和铂用于CO的催化氧化,21-24CO转化率达到100%,所需温度分别约为240和300°C.显然,以MOFs为载体的催化剂催化氧化CO的研究已经取得了重要进展,但较传统载体催化剂还没有明显的优势,其一是MOFs作为载体,其热稳定还有待提高,如在上述载体中,除ZIF-8的分解温度达到500°C外,其余的MOFs载体材料的分解温度均在350°C左右;其二是以MOFs为载体制备的催化剂对CO的催化氧化温度还有待进一步降低.后者不仅是因为高的温度容易使MOFs材料分解从而破坏其结构,而且也为提高其催化剂活性所需要.因此以MOFs作载体的催化剂的研究应该从其热稳定性及催化活性同时提高来进行.注意到Serre研究组25利用Al(NO3)3·9H2O与对苯二甲酸在水热条件下合成了具有立方拓扑结构的金属有机骨架材料MIL-53(Al),MIL-53(Al)是一种三维网络结构的骨架材料,其分子式为Al(OH)[O2C―C6H4―CO2].25这种材料的热稳定性较好,分解温度达到530°C.因而能够适用于温度较高的多相催化反应.
本文以Al(NO3)3·9H2O与对苯二甲酸为原料,采用能促进对苯二甲酸溶解的溶剂热法,在溶剂N,N-二甲基甲酰胺(DMF)中于220°C条件下合成了金属有机骨架材料MIL-53(Al).进而制备了MIL-53(Al)负载的钴催化剂,研究了不同钴含量的催化剂对CO的催化氧化性能,并与Al2O3负载的钴催化剂作了比较.
2 实验部分
2.1 实验试剂
实验所需的试剂:Al(NO3)3·9H2O,对苯二甲酸,N,N-二甲基甲酰胺及Co(NO3)2·6H2O(国药集团化学试剂有限公司,分析纯);三氧化二铝(德国SASOL公司,分析纯);其余试剂均为分析纯,本实验所用的水均为去离子水.
2.2 催化剂的制备
2.2.1 MIL-53(Al)载体的合成
称取3.75 g(0.01 mol)的Al(NO3)3·9H2O和1.66 g(0.01 mol)的对苯二甲酸于小烧杯中,用65 mL DMF溶解,在室温下搅拌0.5 h.将混合好的溶液倒入100 mL聚四氟乙烯内衬的水热反应釜中,于220°C下恒温保持96 h,最后将反应体系自然冷却至室温.将所得的混合液以8000 r·min-1的速率离心,所得固体颗粒分别用DMF(40 mL)和蒸馏水(40 mL)洗涤3次,进行真空抽滤,最后将抽滤的样品放入烘箱干燥12 h,即为MIL-53(Al)载体.
2.2.2 Co/MIL-53(Al)催化剂的合成
称取2.192 g Co(NO3)2·6H2O,用1 mL去离子水溶解后浸渍到4 g MIL-53(Al)载体中,样品通过减压蒸馏去除催化剂中的水分,催化剂在空气中放置12 h后置于风箱中在120°C条件下烘干12 h,最后在400°C下焙烧4 h,即得到质量分数为10%Co/MIL-53(Al)催化剂.按上述相同的方法制备20%Co/MIL-53(Al)和30%Co/MIL-53(Al),催化剂标记为wCo/MIL-53(Al)催化剂(w指不同质量分数的金属Co).wCo/Al2O3催化剂的制备方法同上.
图1为计算所得的Co3O4颗粒在MIL-53(Al)孔道中的分散示意图,由图可见在MIL-53(Al)孔道中的Co3O4颗粒与有机骨架(苯环)几乎不发生作用,发生作用的只有框架节点处的配位氧原子.可以看出Co3O4颗粒几乎是悬吊在分属于两中心Al原子的四个配位氧原子所构成的四边形平面中,因而节点原子对Co3O4颗粒的电子结构影响可以忽略,这种与节点的悬吊型相连方式相比较Co3O4颗粒部分嵌入Al2O3的负载方式,大大增加了Co3O4颗粒的表面积.
2.3 载体和催化剂的表征
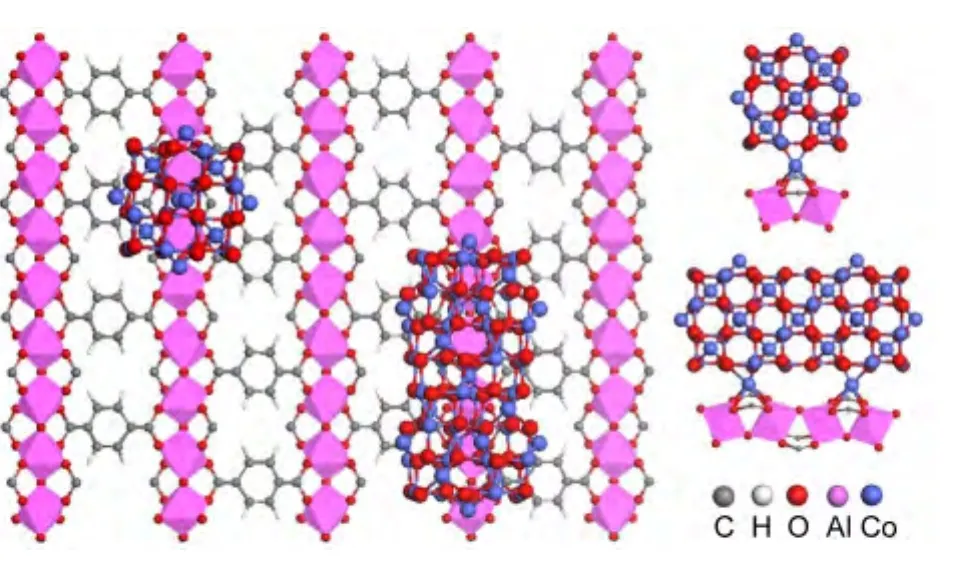
图1 Co3O4纳米颗粒在载体MIL-53(Al)上的分散示意图Fig.1 Schematic diagrams of dispersion of Co3O4 nanoparticles on the MIL-53(Al)carrier
TG-DSC测试是在德国NETZSCH STA449F3型同步热分析仪上进行,样品质量为10 mg,升温速率为10 °C·min-1,O2流量为50 mL·min-1,载体MIL-53(Al)分析前在100°C条件下干燥2 h.FTIR测试在美国NICOLET 8700智能红外光谱仪上进行(4000-400 cm-1).XRD测试在德国Bruker D8型X射线衍射仪上进行,光源为Cu靶Kα线,管电压30 kV,管电流20 mA,扫描速率10(°)·min-1,扫描范围2θ=5°-80°.N2物理吸附-脱附测试在美国MicromeriticsASAP 2020 HD88物理吸附仪上进行的,样品预先在200°C条件下真空脱气3 h,样品的比表面积采用BET法(相对压力p/p0=0.03-0.1间的8个点)计算,孔体积以吸附质相对压力为0.996时的吸附量来计算.TEM是在美国FEI Tecnai G220 S-TWIN透射电子显微镜上进行.氢气程序升温还原(H2-TPR)测试在美国ZETON ALTAMIRA公司的AMI-200型催化剂多功能表征仪上进行,检测器为热导池检测器.首先称取约0.05 g催化剂置于U-型石英反应管中,在氩气(30 mL·min-1)气氛下以10 °C·min-1升温至150°C吹扫1 h,然后将温度降至50°C.再将气体切换为10%H2/Ar(30 mL·min-1)并吹扫至基线平稳.再以10°C·min-1的升温速率从50°C升至500°C,并在500°C下保持半小时,采用热导池检测器记录氢气信号.
2.4 催化剂的CO催化氧化反应
CO的催化氧化活性评价在内径为11 mm的固定床反应器中进行,催化剂的装量为1.0 g,反应气(3%CO和空气)的气体流速为52000 mL·h-1,升温速率为2°C·min-1,反应压力为常压,反应气和尾气的组成由在线的Agilent Micro GC 3000A气相色谱分析.CO的转化率(XCO)根据CO的消耗量来计算:XCO=(VCOa-VCOb)/VCOa×100%,其中VCOa为反应气中CO的初始体积,VCOb为尾气中CO的最终体积.
3 结果与分析
3.1 载体MIL-53(Al)的TG-DSC分析
载体MIL-53(Al)在30-600°C范围内的热分解行为如图2所示.MIL-53(Al)的TG曲线分为2个阶段:第一阶段在100-500°C,此阶段的失重约为26%左右,这可能是合成样品中的水份以及残留的有机物的分解挥发所致,由于吸收与放出的热量相当,相应的DSC曲线吸放热不明显;第二阶段是500°C以后,此阶段失重约为56%,失去的质量是骨架坍塌后对苯二甲酸的燃烧分解.相应的DSC曲线在500°C以后表现出明显的放热趋势,到550°C放热峰达到最大,说明MIL-53(Al)中对苯二甲酸开始分解燃烧并放出热量.载体MIL-53(Al)的TGDSC曲线表明载体在500°C以前的稳定性很好,能够负载金属用于CO的催化氧化反应.
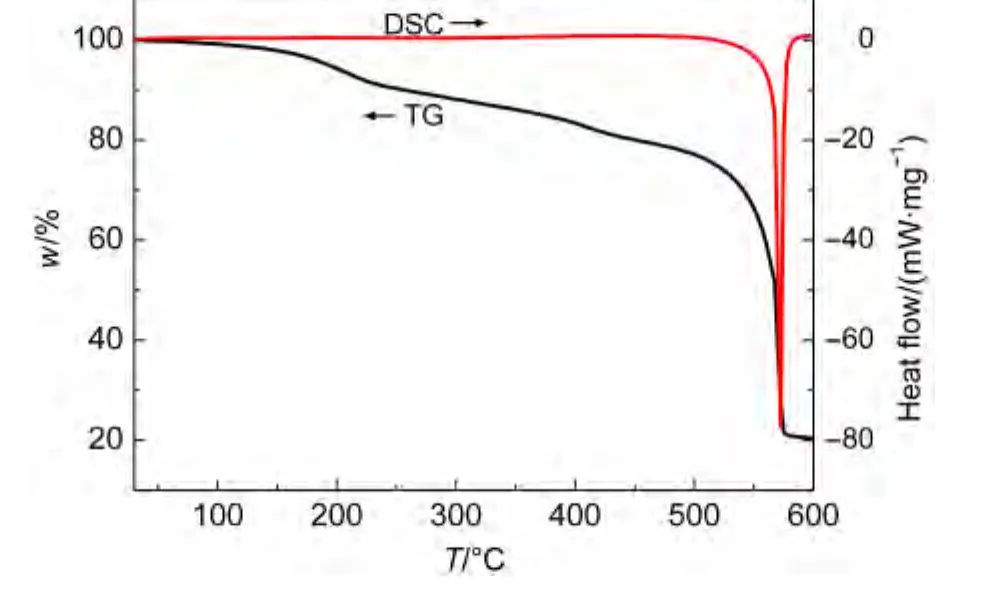
图2 载体MIL-53(Al)的热重-差热扫描量热(TG-DSC)曲线Fig.2 Thermogravimetric-differential scanning calorimeter(TG-DSC)curves of support MIL-53(Al)
3.2 载体MIL-53(Al)的FT-IR分析
图3 是载体MIL-53(Al)焙烧前后的红外光谱图,载体的焙烧温度为450°C,焙烧前后样品的主要特征峰基本保持一致.样品在1690 cm-1附近出现强吸收峰,应归属为―C=O特征振动峰;在1596和1510 cm-1附近的强吸收峰应归属为―(O―C―O)―的反对称伸缩振动吸收峰νasym(CO2-);而在1510和1416 cm-1的强吸收峰应归属为―(O―C―O)―的对称伸缩振动吸收峰νsym(CO2-);在3424 cm-1附近的吸收峰可归属为―OH的伸缩振动吸收峰,这与文献26报道的一致.焙烧前后的红外光谱图基本一致,表明载体MIL-53(Al)在450°C焙烧后结构仍然保持不变,因此Co/MIL-53(Al)系列催化剂在400°C的温度下焙烧不会破坏MIL-53(Al)的结构.
3.3 载体与催化剂的XRD分析
图4 为载体MIL-53(Al)和Al2O3分别负载不同钴负载量的催化剂的XRD图.由文献25可知,MIL-53(Al)的特征衍射峰为 12.3°、16.4°、21.9°和 45.9°.从图中可以看出,用对苯二甲酸和Al(NO3)3·9H2O溶于DMF水热合成的MIL-53(Al)的特征衍射峰的位置和相对强度均与文献25报道的一致.负载钴后的MIL-53(Al)与未负载的MIL-53(Al)相比,其衍射锋强度降低,但在2θ=16.4°,21.9°,45.9°处的特征峰没变,说明MIL-53(Al)负载Co后的基本骨架结构没变.Al2O3的特征衍射峰位于 2θ=37.7°,46.4°,67.3°处,与文献27中χ-Al2O3的特征衍射峰位置一致.在2θ=31.2°,36.8°,45.1°,65.4°处出现Co3O4晶体的衍射峰,表明Co3O4颗粒已经负载到载体上.
3.4 载体和催化剂的N2物理吸附-脱附分析
载体和催化剂的N2物理吸附-脱附等温线如图5所示,在高压部分(p/p0>0.85),曲线(a)氮气吸附量陡增,表明MIL-53(Al)样品中存在一定的中孔结构,样品的中孔孔径较大,这些较大中孔来自于纳米颗粒堆积而成的间隙孔.在高压部分(p/p0>0.85),曲线(e)氮气吸附量也略有增加,表明Al2O3样品中也存在一定的中孔结构,相比于MIL-53(Al)样品中的中孔,Al2O3样品中的中孔孔径略小一些.采用BET方法计算得到载体MIL-53(Al)的比表面积远高于Al2O3的比表面积.两种载体负载钴后,相应的比表面积均减小,结果见表1.
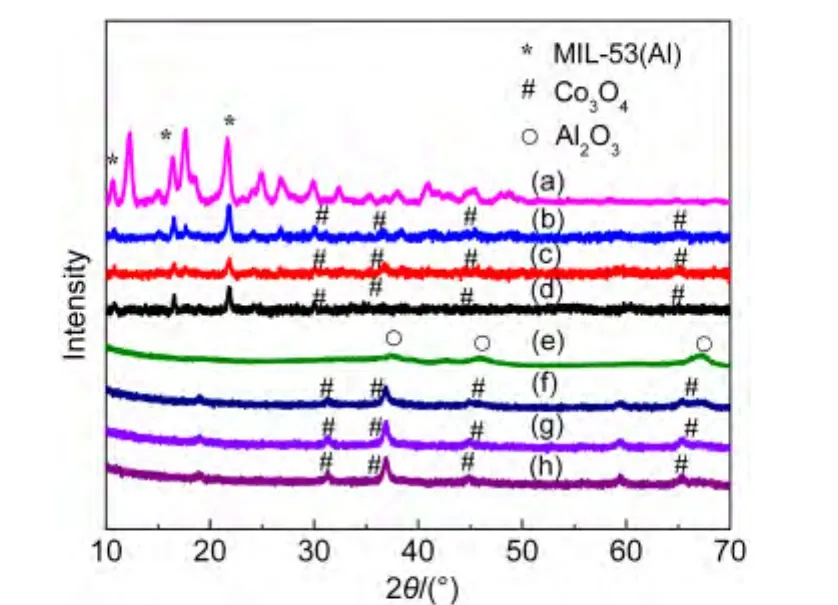
图4 载体和催化剂的XRD图Fig.4 XRD patterns of support and catalysts

图5 载体MIL-53(Al),Al2O3及其负载的钴催化剂的N2物理吸附-脱附等温线Fig.5 N2adsorption-desorption isotherms of the supports MIL-53(Al),Al2O3,and the corresponding catalysts
催化剂的N2物理吸附-脱附数据见表1.由表可见,载体MIL-53(Al)的比表面积为927.4 m2·g-1,孔容为1.43 cm3·g-1;而载体Al2O3的比表面积为190.4 m2·g-1,孔容为 0.53 cm3·g-1.显然载体 MIL-53(Al)比载体Al2O3的比表面积高出许多,比表面积的增加有利于更好地分散催化剂.由XRD计算结果可知,载体MIL-53(Al)上Co3O4纳米颗粒的粒径更小一些.在负载型催化剂上,金属颗粒的分散度和颗粒的大小对CO吸附以及活化起到了关键作用,随着金属颗粒的减小和分散度的提高,其催化活性也将提高.3由表1可知MIL-53(Al)和Al2O3负载钴后,其比表面积和孔容均变小,且随着负载量的增多,比表面积逐渐减小,说明Co3O4纳米颗粒分别进入了载体的孔道中(如图1所示).
3.5 载体和催化剂的TEM分析
图6是载体MIL-53(Al)和Al2O3的TEM照片,从图6(a)可以看出,载体MIL-53(Al)在450°C焙烧后,样品大部分以长条形为主,长度约为100 nm左右,颗粒之间均匀分散.从图6(b)可以看出,载体Al2O3在450°C焙烧后,样品粒径大小不一,类似针状颗粒并形成聚集体,从形貌上分析,载体MIL-53(Al)的形状更均匀,颗粒更小且颗粒之间分散更为均匀.
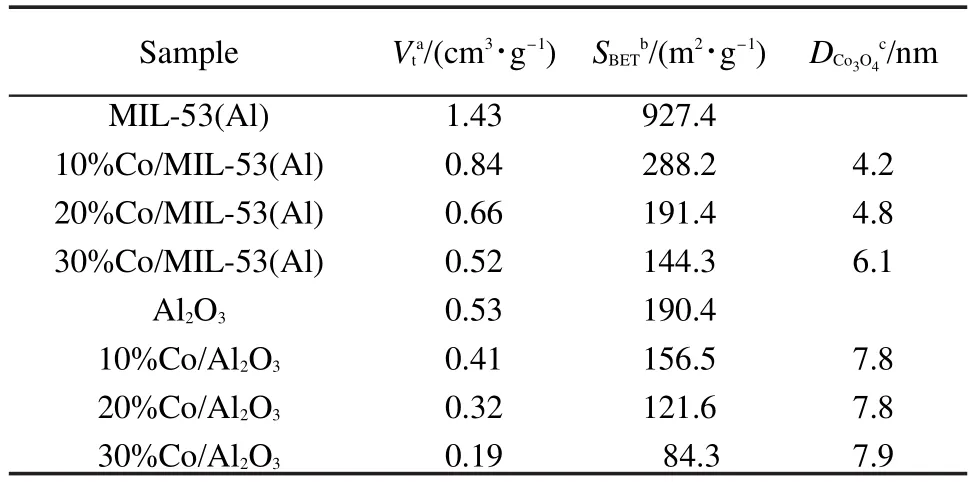
表1 催化剂的N2吸附-脱附数据Table1 N2adsorption-desorption data of catalysts
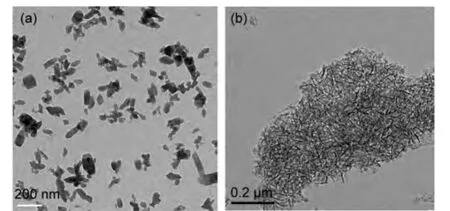
图6 载体MIL-53(Al)(a)和Al2O3(b)的透射电镜(TEM)照片Fig.6 Transmission electron microscopy(TEM)images of supports MIL-53(Al)(a)andAl2O3(b)
图7(a,b,c)是催化剂(10%,20%,30%)Co/MIL-53(Al)的TEM照片,图7(d,e,f)是催化剂(10%,20%,30%)Co/Al2O3的TEM照片.从图7(a,b,c)可知Co3O4颗粒在MIL-53(Al)的表面上分散的颗粒相对较小.从图7(d,e,f)可知在载体Al2O3上的Co3O4颗粒是一种没有规则形状的钴物种团聚体.与Co3O4颗粒部分嵌入载体Al2O3的负载方式相比,MIL-53(Al)载体上的Co3O4颗粒是与分属于两中心Al原子的四个配位节点的氧原子以悬吊性方式相连(见图1),这种悬吊性相连方式使Co3O4颗粒的分散性更好.又由于框架配合物中Co3O4颗粒的沉积位点空间分布均匀,这有利于Co3O4在三维框架内“均匀”成核、长大,从而获得分散较均匀的更小颗粒.因此载体MIL-53(Al)上负载的Co3O4颗粒更细小,分散性更好.
3.6 催化剂的H2-TPR分析
图8是不同催化剂的H2-TPR谱图.其中图8(a,b,c)分别是(30%,20%,10%)Co/MIL-53(Al)催化剂的H2-TPR图,图8(d,e,f)分别是(30%,20%,10%)Co/Al2O3催化剂的H2-TPR图.对于MIL-53(Al)负载的钴基催化剂,(10%,20%,30%)Co/MIL-53(Al)第一步还原的温度分别为285、291和305°C,对应的是Co3O4到CoO的还原过程;第二步还原温度分别为329、348和374 °C,对应的是CoO到Co0的还原过程.对于Al2O3负载的钴基催化剂,(10%,20%,30%)Co/Al2O3第一步还原温度在402、403和410 °C,对应的是Co3O4到CoO的还原过程;第二步还原温度在500°C以上,对应的是CoO到Co0的还原过程.由图可知,两种催化剂随着钴含量从10%到30%的增加,每一步的还原温度都略有增加,这是高的钴负载量导致体相四氧化三钴量增加,体相氧化物的还原温度比分散态的还原温度高.另外,从图中可以看出Co/MIL-53(Al)催化剂两步还原的温度均低于Co/Al2O3催化剂的还原温度,H2-TPR的还原温度的高低可以表征催化剂中氧的活性大小,28,29一般H2-TPR的还原温度越低,越容易还原,则对CO的氧化活性越好.结合Co3O4颗粒在MIL-53(Al)载体上的分散示意图(如图1所示)分析可知,相比于Al2O3颗粒,MIL-53(Al)中由于苯环的存在,MIL-53(Al)中吸附Co3O4的氧参与了苯环中的共轭,消弱了金属与载体之间的作用,这种作用有利于钴氧化物的还原,使得Co/MIL-53(Al)催化剂的还原温度降低,从而有利于催化反应.
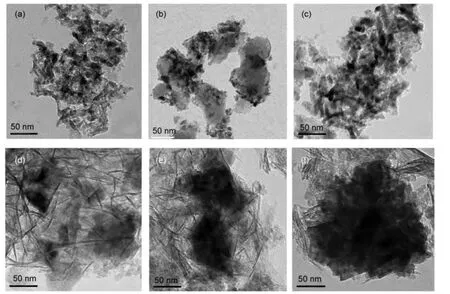
图7 不同催化剂的TEM照片Fig.7 TEM images of different catalysts

图8 不同催化剂的氢气程序升温还原(H2-TPR)谱图Fig.8 Hydrogen temperature-programmed reduction(H2-TPR)profiles of different catalysts
3.7 催化剂的CO氧化性能
MIL-53(Al)负载不同含量的Co3O4纳米颗粒用于CO的催化氧化反应,其转化率随反应温度变化曲线如图9(a,b,c)所示.从图中可以看出,随着负载量的增大,其转化率达到100%所需的温度先降低后增加.10%Co/MIl-53(Al)使CO转化率达到99%时所需温度为240°C;20%Co/MIl-53(Al)与30%Co/MIl-53(Al)使CO转化率达到100%时所需温度分别为180与200°C,说明负载量超过一定程度时,过多极细小的Co3O4颗粒分散在载体MIl-53(Al)的孔道中使Co3O4的分散度有所下降.因此,在此实验中MIl-53(Al)对钴的最佳负载量大约为20%.在相同的条件下,Al2O3负载钴催化剂用于CO的催化氧化时,其转化率随反应温度变化曲线如图9(d,e,f)所示.从图中可以看出,10%Co/Al2O3使CO的转化率达到100%时所需的温度为315°C,20%Co/Al2O3和30%Co/Al2O3使CO的转化率达到100%时所需的温度分别为300和255°C.在相同条件下,MIL-53(Al)作为载体使CO转化率达到100%所需的温度均低于Al2O3负载钴催化剂.同时,在CO催化氧化反应中,MIL-53(Al)负载钴催化剂优于目前金属有机骨架材料ZIF-8作为载体负载的贵金属铂催化剂的催化性能.20从上述比较中可以看出,不同载体MIL-53(Al)和Al2O3负载的钴催化剂均采用同样负载方式负载等量的活性组分Co3O4颗粒却表现出不同的催化氧化活性,结合比表面积数据分析可知,CO的催化活性与载体的比表面积大小有关,比表面积越大,越有利于活性组分的分散.同时结合Co3O4颗粒在MIL-53(Al)载体上的分散示意图(见图1)分析可知,在载体MIL-53(Al)中Co3O4颗粒与节点氧原子的相连方式使MIL-53(Al)吸附Co3O4的氧位点增多,增加了其吸附位,从而增加了供氧数目,供氧数目越多催化剂的供氧能力越强,则对CO的氧化活性越好.又由于CO的催化活性与Co3O4颗粒的粒径大小有关,已有文献30,31报道催化剂颗粒的粒径越小,催化活性越好.结合实验中的TEM结果和XRD数据可知,载体MIL-53(Al)负载的Co3O4颗粒的粒径比载体Al2O3负载的Co3O4颗粒的粒径小,因此MIL-53(Al)负载钴催化剂的活性更好.
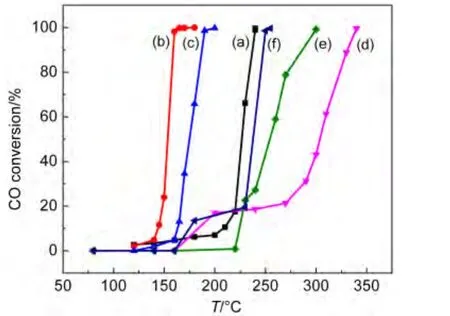
图9 不同催化剂的CO催化氧化活性对比图Fig.9 Catalytic performances of different catalysts for CO oxidation
4 结论
采用溶剂法合成了热稳定性高的金属有机骨架材料MIL-53(Al),用此材料负载钴催化剂用于CO的催化氧化反应,并与Al2O3负载的钴催化剂进行了对比.采用热重分析、傅里叶变换红外光谱、X射线衍射、N2物理吸附-脱附、透射电镜、氢气程序升温还原等方法对催化剂的结构性质进行了表征.结果表明,以热稳定性高,比表面积大的MIL-53(Al)为载体负载的钴催化剂用于CO的催化氧化反应,其催化活性不仅明显高于Al2O3负载的钴催化剂,而且相较现有文献报道的以MOFs为载体制备的CO的催化剂,其热稳定性及催化活性均有明显提高.
致谢: 本文实验得到了中南民族大学催化材料科学湖北省暨国家民委-教育部共建重点实验室的支持!感谢重点实验室的老师和同学对做CO催化反应给予的帮助!感谢中国地质大学可持续能源实验室的韩波老师和冀转同学的帮助!
(1) Gong,Y.;Chen,H.R.;Cui,X.Z.;Jiang,W.;Shi,J.L.J.Inorg.Mater.2013,28,992.[龚 云,陈航榕,崔香枝,江 莞,施剑林.无机材料学报,2013,28,992.]doi:10.3724/SP.J.1077.2013.12711
(2) Prasad,R.;Singh,P.Catal.Rev.2012,54,224.doi:10.1080/01614940.2012.648494
(3) Zhan,J.;Chen,J.;Huang,X.S.;Li,G.S.Prog.Chem.2012,24,1245.[张 俊,陈 娟,黄新松,李广社.化学进展,2012,24,1245.]
(4) Liu,Y.L.;You,C.Y.;Li,Y.;He,T.;Zhang,X.Q.;Suo,Z.H.Acta Phys.-Chim.Sin.2010,26,2455.[刘玉良,由翠英,李杨,何 涛,张香芹,索掌怀.物理化学学报,2010,26,2455.]doi:10.3866/PKU.WHXB20100909
(5)Wen,L.;Lin,Z.Y.;Zhou,J.Z.;Gu,P.Y.;Fu,J.K.;Lin,Z.H.Acta Phys.-Chim.Sin.2008,24,581.[文 莉,林种玉,周剑章,古萍英,傅锦坤,林仲华.物理化学学报,2008,24,581.]doi:10.3866/PKU.WHXB20080407
(6) Sun,J.F.;Ge,C.Y.;Yao,X.J.;Cao,Y.;Zhang,L.;Tang,C.J.;Dong,L.Acta Phys.-Chim.Sin.2013,29,2451.[孙敬方,葛成艳,姚小江,曹 原,张 雷,汤常金,董 林.物理化学学报,2013,29,2451.]doi:10.3866/PKU.WHXB201309041
(7) Gulari,E.;Guldur,C.;Srivannavit,S.;Osuwan,S.Appl.Catal.A:Gen.1999,182,147.doi:10.1016/S0926-860X(99)00002-2
(8) Ferey,G.;Mellot-Draznieks,C.;Serre,C.;Millange,F.;Dutour,J.;Surble,S.;Margiolaki,I.Science 2005,309,2040.doi:10.1126/science.1116275
(9) Li,H.;Eddaoudl,M.;O'keeffe,M.;Yaghl,O.M.Nature 1999,402,276.doi:10.1038/46248
(10)Chae,H.K.;Siberio-Pérez,D.Y.;Kim,J.;Go,Y.B.;Eddaoudi,M.;Matzger,A.J.;O'Keeffe,M.;Yaghi,O.M.Nature 2004,6974,523.
(11)Chen,B.;Ockwig,N.W.;Millward,A.R.;Contreras,D.S.;Yaghi,O.M.Angew.Chem.Int.Edit.2005,44,4745.
(12)Corma,A.;Garcia,H.;Llabresi,Xamena,F.X.Chem.Rev.2010,110,4606.doi:10.1021/cr9003924
(13) Seo,J.S.;Whang,D.;Lee,H.;Jun,S.I.;Oh,J.;Jeon,Y.J.;Kim,K.Nature 2000,404,982.doi:10.1038/35010088
(14) Banerjee,M.;Das,S.;Yoon,M.;Choi,H.J.;Hyun,M.H.;Park,S.M.;Seo,G.;Kim,K.J.Am.Chem.Soc.2009,131,7524.doi:10.1021/ja901440g
(15) Combelles,C.;Yahia,M.B.;Pedesseau,L.Phys.Chem.2010,114,9518.
(16) Demir-Cakan,R.;Morcrette,M.;Nouar,F.;Davoisne,C.;Devic,T.;Gonbeau,D.;Dominko,R.;Serre,C.;Ferey,G.;Tarascon,J.M.J.Am.Chem.Soc.2011,133,16154.doi:10.1021/ja2062659
(17)Li,Y.F.;Wei,M.D.Mater.Chem.2011,21,17259.doi:10.1039/c1jm12754c
(18) Zou,R.Q.;Sakurai,H.;Xu,Q.Angew.Chem.Int.Edit.2006,45,2542.
(19)Zou,R.Q.;Sakurai,H.;Han,S.;Zhong,R.Q.;Xu,Q.J.Am.Chem.Soc.2007,129,8402.doi:10.1021/ja071662s
(20) Jiang,H.L.;Liu,B.;Akita,T.;Haruta,M.;Sakurai,H.;Xu,Q.J.Am.Chem.Soc.2009,131,11302.doi:10.1021/ja9047653
(21)Zhang,F.;Chen,C.;Xiao,W.M.;Xu,L.;Zhang,N.Catal.Commun.2012,26,25.doi:10.1016/j.catcom.2012.04.028
(22)Zamaro,J.M.;Perez,N.C.;Miro,E.E.;Casado,C.;Seoane,B.;Tellez,C.;Coronas,J.Chem.Eng.J.2012,180,195.
(23) Ramos-Fernandez,E.V.;Pieters,C.;Linden,B.V.;Juan-Alcañiz,J.;Serra-Crespo,P.;Verhoeven,M.W.G.M.;Niemantsverdriet,H.;Gascon,J.;Kapteijn,F.J.Catal.2012,289,42.doi:10.1016/j.jcat.2012.01.013
(24)Aijaz,A.;Karkamkar,A.;Choi,Y.J.;Tsumori,N.;Nonnebro,E.;Autrey,T.;Shioyama,H.;Xu,Q.J.Am.Chem.Soc.2012,134,13926.doi:10.1021/ja3043905
(25) Loiseau,T.;Serre,C.;Huguenard,C.;Fink,G.;Taulelle,F.;Henry,M.;Bataille,T.;Ferey,G.Chem.Eur.J.2004,10,1373.
(26) Zhang,J.;Liu,S.S.;Song,L.F.;Jiang,C.H.;Jiao,C.L.;Wang,S.;Zhang,Y.;Zhao,J.N.;Gao,X.Y.;Xu,F.;Sun,L.X.Materials China 2009,28,28.[张 箭,刘淑生,宋莉芳,姜春红,焦成丽,王 爽,张 耀,赵军宁,高秀英,徐 芬,孙立贤.中国材料进展,2009,28,28.]
(27) Li,B.;Shao,L.L.Inorg.Chem.Indus.2008,40,54.[李 波,邵玲玲.无机盐工业,2008,40,54.]
(28)Zhu,B.;Luo,M.F.;Chen,P.;Zhou,L.H.;Yuan,X.X.;Wu,H.L.J.Fuel Chem.Technol.1997,25,32.[朱 波,罗孟飞,陈 平,周烈华,袁贤鑫,吴红丽.燃料化学学报,1997,25,32.]
(29) Zhou,R.X.;Jiang,X.Y.;Mao,J.X.;Zheng,X.M.Chin.J.Catal.1997,18,53.[周仁贤,蒋晓原,毛建新,郑小明.催化学报,1997,18,53.]
(30)Haruta,M.;Tsubota,S.;Kobayashi,T.;Kageyama,H.;Genet,M.J.;Delmon,B.J.Catal.1993,144,175.doi:10.1006/jcat.1993.1322
(31)Jia,M.J.;Zhang,W.X.;Tao,Y.G.;Wang,G.Y.;Cui,X.H.;Zhang,C.L.;Wu,T.H.;Dong,G.Q.;Li,X.M.Chem.J.Chin.Univ.1999,20,637.[贾明君,张文祥,陶玉国,王桂英,崔湘浩,张春雷,吴通好,董国强,李雪梅.高等学校化学学报,1999,20,637.]
猜你喜欢
华人时刊(2022年9期)2022-09-06
机械工业标准化与质量(2022年6期)2022-08-12
小学生学习指导(高年级)(2022年3期)2022-03-29
华人时刊(2020年15期)2020-12-14
小学生导刊(高年级)(2017年2期)2017-06-10
中国调味品(2017年2期)2017-03-20
小学生导刊(2017年6期)2017-02-10
小学生导刊(高年级)(2016年1期)2016-01-29
广州大学学报(自然科学版)(2015年4期)2015-12-23
中学化学(2015年2期)2015-06-05
