
囊性纤维化对呼吸系统影响的队列研究系统评价
2014-08-30 06:51曾晓露熊鸿燕叶诗洋王太武
中华肺部疾病杂志(电子版) 2014年4期
曾晓露 熊鸿燕 叶诗洋 王太武
囊性纤维化(cystic fibrosis, CF)是一种外分泌腺功能紊乱的常染色体隐性遗传性疾病,主要由囊性纤维化跨膜传导调节因子(cystic fibrosis transmembrane conductance regulator, CFTR)基因突变引起[1]。该病可累及呼吸、消化、内分泌、生殖等多个系统,其临床表现随年龄的不同而有所不同,新生儿期主要表现为胎粪性肠梗阻、持续黄疸、肠闭锁等;婴儿期主要为发育不良、金黄色葡萄球菌肺炎等;儿童期主要表现为远端肠梗阻、慢性鼻窦炎或鼻息肉、慢性胰腺炎、肝病等;青年期和成年期主要为支气管扩张、咯血、门静脉高压症、变应性支气管肺曲霉病、无精症等[2]。除此之外,在任何年龄都可能出现杵状指、咸味皮肤、咳嗽、咳痰、呼吸道分泌物粘性铜绿假单胞菌的分离、低氯性代谢性碱中毒等。其中反复急进性的肺部感染为其主要的致死原因[3]。
据报道,该病好发于欧美白色人种,发病率约为1/3000[4]。欧美国家早在1985年就开始对CF实行新生儿筛查,在美国CF基金会(US CFF)、欧洲CF协会(ECFS)等组织的推动下,现今已在美国、英国等国家实现了全面的新生儿筛查,建立了多中心的观察队列,这对于CF患者的早发现和早治疗起到了积极作用。相反的,在非洲及亚裔人群中,CF被认为极为“罕见”,仅日本有报道CF的估计患病率为1/350 000,但这一数据是根据日本的病例报告和同时期新生儿出生率估算得到,准确性不得而知[5-6]。在中国,由于新生儿CF筛查实验及汗液实验未普及,大陆地区从1975年至今,除去资料不完整或缺乏确切诊断证据者外,仅报道26例CF患者[7]。但CF相关的临床症状,在中国人群中并不罕见,例如新生儿的胎粪性肠梗阻,儿童的慢性胰腺炎,肝病,成年人的支气管扩张,无精症等,这些疾病有很大一部分病因都不明确,是否与CF有关,已有研究者对此提出了疑问,但准确结论还有待进一步研究[8-9]。
本研究基于欧美国家已对CF建立了多个大样本的研究队列,且队列研究为观察性研究中论证强度最高的研究方法,故选取CF的队列研究进行系统评价;同时因为反复肺部感染导致的肺功能不全是80%以上的CF患者的死亡原因[3],故本次系统评价主要针对呼吸系统进行,旨在对近5年CF在世界范围报道病例的分布情况、临床症状、肺部细菌的感染及耐药情况做一描述,从而提高在CF研究尚不足地区的临床医生对该病的认识,帮助世界各地CF患者及早认识发觉此病,并展开合理治疗,提高生存率和生活质量。
资料与方法
一、检索策略
选择PubMed、Medline(via OvidSP)、Embase、Web of Science和the Cochrane Database 5个数据库进行检索(检索时间为2008年5月至2013年5月)。检索的主题词为:“cystic fibrosis”, “cohort study”; 限制条件为:“published in the last 5 years”, “humans”。用Endnote软件对检索到的文献进行管理并去重。
二、纳入、排除标准
在文献纳入过程中,由2位研究者独立进行审核,如有异议的,由2位研究者讨论决定纳入与否,以保证研究质量。
根据研究类型与疾病特点,文献纳入与排除标准从文献类别、样本量、诊断标准等方面确定:
1. 研究类型:队列研究,或者所使用的患者资料为前瞻性收集的观察性研究;排除综述、病例报告、或动物实验。
2. 文中病例的样本量大于等于20例。
3. 文中CF有明确的诊断标准、确诊方法或数据来源于以前已发表的CF队列数据库。
4. 排除非英文的文献。
三、资料提取及分析
由2位研究者按照设计好的提取表格独立进行资料提取,内容包括:发表的国家、年限、发表病例数、病例性别与年龄、研究类型、CF临床特征、CF肺部疾病的相关症状、CF肺部感染的相关细菌及耐药情况。
运用EXCEL2007对提取的信息进行储存分析,并运用ArcGIS10.0软件进行作图。
结 果
一、文献检索结果
初步检索出584篇相关文献,通过阅读文章题目、摘要排除179篇文献,剩余文献阅读全文,按照纳入排除标准进行筛选,最终纳入260篇文献,见图1。
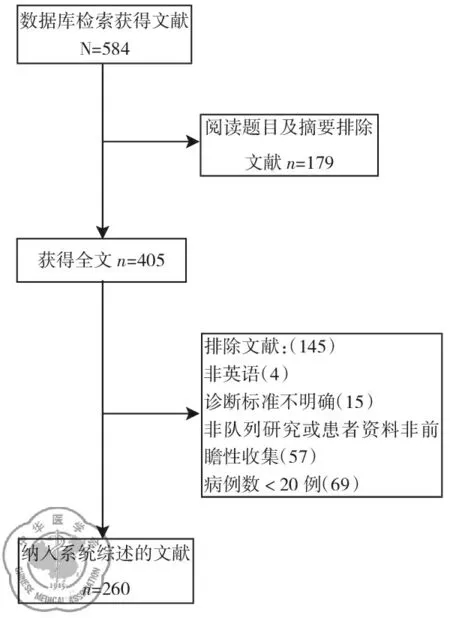
图1 文献纳入排除流程图
二、病例基本特征及发表文献的分布
本次系统评价一共纳入的260篇文献,总共包含260 879例CF患者病例(由于一些文献的病例来自于同一个队列数据库,而他们的纳入排除标准又有部分重叠,不能进行区分,故本次研究提及的病例数为发表病例数,并不是绝对病例数)。这些病例的年龄2个月~63岁,平均年龄18.7岁;男女比例为1.05︰1(95 876男/90 756女)。由于有些文献只给出了病例的平均年龄和年龄范围,我们没能得到病例的年龄分布数据。
CF在多个国家均有报道,按照方法中的纳入标准,本研究选取了近5年CF队列研究且病例数大于20的文献,以保证病历资料的准确性和代表性,最后纳入的260篇文献,分别来自于21个国家(图2)。其中86篇文献218 679病例来自于美国,37篇文献15 436病例来自于英国,20篇文献7825病例来自于加拿大,24篇文献2700病例来自于法国,18篇文献2341病例来自于澳大利亚,13篇文献3235病例来自于德国,11篇文献3324病例和1119病例分别来自于意大利和荷兰。
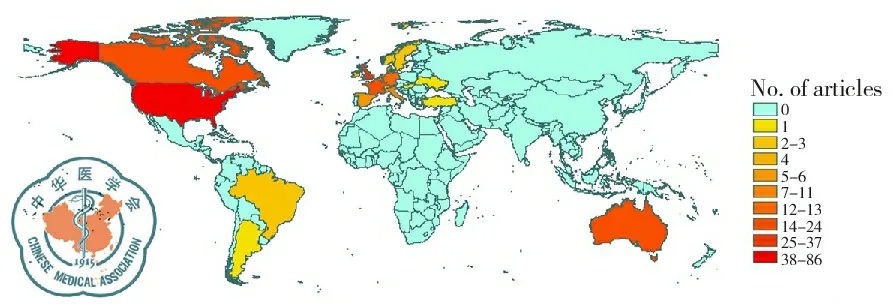
图2 260篇发表文献的世界分布情况
三、诊断标准及临床表现
CF是一种多器官的全身性疾病,临床表现变异很大,不同年龄、不同个体表现都可能不同,这对CF的诊断造成了一些困难。美国CF基金会、欧洲CF协会、CFF协作组和WHO都发表有相应的诊断指南[10-12]。
在本次的系统评价中,综合各种诊断标准,我们使用的诊断标准如下:有1个或多个CF的典型临床表现(胰腺或汗腺功能异常、呼吸道疾病、无精症等),合并2次以上汗液实验阳性(汗液氯离子浓度>60 mmol/L),或者合并2个致病性CFTR突变位点。
由此诊断标准纳入文献后,归纳出病例的主要临床表现为以下4个方面:胃肠道症状、呼吸系统症状、内分泌疾病和生殖系统疾病,见表1。具体的151 948例 (58.24%)表现出CF肺病,其中最主要的是慢性呼吸道感染,其他还包括支气管扩张、肺不张、呼吸衰竭等;3 3185例 (12.72%)患有胰腺功能不全;3938例 (1.51%)患有CF相关的糖尿病;2389例 (0.92%)患有CF相关的肝病,特别是胆汁性肝硬化;2205例 (0.85%)患有胎粪性肠梗阻(病例都为婴儿期患此病);1136例(0.44%)患有鼻窦疾病,包括鼻窦炎和鼻息肉;92例(0.04%)患有无精症。CF的相关症状除以上一些外,还有许多,例如骨质疏松、慢性腹泻、营养不良等症状在一些文献中也有叙及,因病例数太少,未做统计。
注:CF:囊性纤维化
四、CF肺部常见症状
对260篇文献中提及肺部症状的22篇文献进行总结,发现CF肺部常见症状依次为:细菌持续定植于气道、慢性咳嗽咳痰、喘息、胸闷、呼吸困难、咯血、发绀、杵状指。实验室检查多表现为FEV1下降和SaO2下降。影像学检查多表现为支气管扩张、肺浸润影和空气陷闭征,见表2。

表2 CF肺病报道的主要症状、体征、实验室检查
注:CF:囊性纤维化
五、肺部细菌感染及耐药情况
本次系统评价显示,CF相关的肺部病原菌主要包括铜绿假单胞菌(pseudomonasaeruginosa, PA)、金黄色葡萄球菌(staphylococcusaureus, SA)、耐甲氧西林金黄色葡萄球菌(methicillin-resistantstaphylococcusaureus, MRSA)、洋葱伯克杆菌属(burkholderiacepacia, BC)、嗜麦芽寡养单胞菌(stenotrophomonasmaltophilia, SM)、流感嗜血杆菌(haemophilusinfluenza, HI)、烟曲霉菌(aspergillusfumigatus, ASP)、氧化木糖无色杆菌(achromobacterxylosoxidans, AX)和肺结核分支杆菌(nontuberculousmycobacteria, NTM)。其中PA和SA为最主要的病原菌,分别以97 156例和49 578例占总病例数的37.24%和19.00%;MRSA占2.67%。ASP是本次统计的唯一真菌,感染病例占总病例的1.01%。其他格兰阴性杆菌——BC、SM、HI、AX、NTM分别占到1.81%、1.18%、1.38%、0.66%和0.02%。PA对多种抗生素耐药,包括阿米卡星、 氨曲南、 头孢吡肟 、孢他啶、环丙沙星 、粘菌素、美罗培南、庆大霉素、亚胺培南、氧氟沙星、哌拉西林、他唑巴坦、替卡西林-克拉维酸;SA主要对阿奇霉素耐药;而MRSA对环丙沙星、克林霉素、红霉素、庆大霉素、利福平都耐药;ASP对抗真菌药伊曲康唑产生耐药,见表3。
注:“+”为病原菌有耐药情况,“-”为病原菌没有耐药情况
讨 论
本研究所作系统评价显示,在260篇文献关于囊性纤维化的队列研究中,发表报道前5位的国家依次为美国、英国、加拿大、意大利和法国,一方面可能由于在这些国家CFTR突变基因携带人群数量大,另一方面可能由于这些地区的人们对CF的重视程度高,国家资金投入大,对CF的研究工作处于国际先进水平,诊断治疗技术都处于领先地位,有利于CF的检出。
CF的发表文献在各大洲的分布是不平衡的,在统计出的21个国家中,除以色列为亚洲国家外,其余都为欧美国家。发表病例数在这些国家中的分布也是不平衡的,美国的发表病例数占总发表病例数的84%,居于第一位。这可能是因为美国为开展新生儿筛查最早的国家,CF队列建立和病案管理较为全面,这也说明了对CF的重视与投入程度,直接影响着CF的检出率。
CF的临床表现以肺部疾病为主,特别是反复进行性的肺部感染,已被证明是CF的主要致死原因[3]。从结果可以看出,CF肺部病原菌的感染情况与院内肺部感染的细菌分布十分相似,都以革兰阴性杆菌为主[13-14],特别是铜绿假单胞菌,已被多位学者证明为CF病原菌中感染最广,对治疗和预后影响最大的细菌[15-17]。这种相似的细菌分布,不仅不利于CF肺部感染患者的早期检出,也容易使得临床上按照一般肺部感染进行常规治疗,导致这些患者常年反复住院而得不到准确的诊断。同时,反复的抗生素运用致使耐药性的产生,增大了治疗的难度。这一情况,应引起临床医生的重视。
中国目前没有一篇CF符合病例数大于20人的队列研究,但有学者指出,从1974年到2006年共检索出26例中国CF病例,CF发病率虽然很低,但突变等位基因的绝对量可能相当大[7]。李慧萍等[8]也指出由于亚裔CF患者临床表现没有欧美及高加索人典型,多因支气管扩张、反复肺部感染,如PA感染等久治不愈,或由于近亲婚配而其子女出现典型临床表现时,才引起重视,且亚裔CFTR基因突变谱不同于欧美及高加索人种,因此,可能构成不同于欧美及高加索人种的大量潜在的原发突变和CF患者群。因此在包括中国在内的CF被认为极为“罕见”的亚非国家中,仍不能忽视CF的潜在威胁,应尽早开展早期筛查,建立CF队列,进行CF的研究。
本次系统评价存在着一些局限性。首先,本研究的检索主题词只设定为CF和队列研究,虽提高了研究资料的准确性和验证强度,但研究内容的异质性十分大,使得一些资料不能进行合并,丢失了信息。其次,在一些文献中,对临床症状和肺部病原菌的描述并不全面,例如一些主要内容不是研究病原菌的文献,只给出了最主要的一两种细菌的感染情况,而其他细菌是否感染,并未说明,这可能会导致研究结果的不准确性。最后,由于欧美国家队列研究所纳入的病例,多来自于各个中心或医院的队列数据库,为避免病例的过多重复纳入,本研究只选择了近5年的文献。同时,为了提高病例的代表性,也只纳入了病例数大于20的文献,这使得一些小样本的研究未被纳入。然而一些对CF研究起步较晚的国家,特别是一些亚非国家,还没有建立CF队列,仅有小样本研究资料或病例报告[18-19]。所以,本次系统评价得到的CF发表文献分布信息,可能只代表了CF研究相对领先的一些国家的情况。而对于小样本的CF研究资料,尚有待进一步的评价研究。
参 考 文 献
1 Rommens JM, Iannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping[J]. Science,1989, 245(4922): 1059-1065.
2 O′Sullivan BP, Freedman SD. Cystic fibrosis[J]. Lancet, 373(9678): 1891-1904.
3 Davis PB. Cystic fibrosis since 1938[J]. Am J Respir Crit Care Med, 2006, 173(5): 475-482.
4 Walters S, Mehta A. Epidemiology of cystic fibrosis[M]. In: Hodson M, Geddes DM, Bush A, eds. Cystic fibrosis, 3rd edn. London: Edward Arnold Ltd, 2007: 21-45.
5 陈柏华, 杨 元, 张思仲. 囊性纤维化的分子遗传学研究进展[J]. 中华医学遗传学杂志,1997, 14(4): 243-247.
6 Yamashiro Y, Shimizu T, Oguchi S, et al. The estimated incidence of cystic fibrosis in Japan[J]. J Pediatr Gastroenterol Nutr,1997, 24(5): 544-547.
7 刘亭威, 康 健. 中国人囊性纤维化临床特点分析[J]. 中国全科医学, 2012, 15(24): 2807-2810.
8 李惠萍, 何国钧. 囊性肺纤维化与支气管扩张症的关系以及在中国人群中的研究价值[J]. 中华结核和呼吸杂志,2004, 27(9): 627-629.
9 李 楠, 何 冰, 王广发,等. 囊性纤维化一例报告并文献复习[J]. 中华结核和呼吸杂志,2003, 26(9): 559-562.
10 De Boeck K, Wilschanski M, Castellani C, et al. Cystic fibrosis: terminology and diagnostic algorithms[J]. Thorax,2006, 61(7): 627-635.
11 Farrell PM, Rosenstein BJ, White TB, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report[J]. J Pediatr, 2008, 153(2): S4-S14.
12 Ooi CY, Dupuis A, Ellis L, et al. Comparing the American and European diagnostic guidelines for cystic fibrosis: same disease, different language? [J]. Thorax, 2012, 67(7): 618-624.
13 Schaberg DR, Culver DH, Gaynes RP. Major trends in the microbial etiology of nosocomial infection[J]. Am J Med, 1991, 91(3B): 72S-75S.
14 Millar FA, Simmonds NJ, Hodson ME. Trends in pathogens colonising the respiratory tract of adult patients with cystic fibrosis, 1985-2005[J]. J Cyst Fibros, 2009, 8(6): 386-391.
15 Govan JR, Brown AR, Jones AM. Evolving epidemiology of Pseudomonas aeruginosa and the Burkholderia cepacia complex in cystic fibrosis lung infection[J]. Future Medicine, 2007, 2(2):153-164.
16 Murray TS, Egan M, Kazmierczak BI. Pseudomonas aeruginosa chronic colonization in cystic fibrosis patients[J]. Curr Opin Pediatr, 2007, 19(1): 83-88.
17 Emerson J, Rosenfeld M, McNamara S, et al. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis[J]. Pediatr Pulmonol, 2002, 34(2): 91-100.
18 Nakakuki M, Fujiki K, Yamamoto A, et al. Detection of a large heterozygous deletion and a splicing defect in the CFTR transcripts from nasal swab of a Japanese case of cystic fibrosis[J]. J Hum Genet, 2012, 57(7): 427-433.
19 Uppaluri L, England S, Scanlin T. Clinical evidence that V456A is a Cystic Fibrosis causing mutation in South Asians[J]. J Cyst Fibros, 2012, 11(4): 312-315.
猜你喜欢
传染病信息(2022年3期)2022-07-15
世界科学技术-中医药现代化(2022年2期)2022-05-25
中国药学药品知识仓库(2021年18期)2021-02-28
科学导报·学术(2020年26期)2020-10-21
小学生学习指导(低年级)(2020年4期)2020-06-02
软件(2020年3期)2020-04-20
中国中医急症(2019年10期)2019-05-21
中国临床医学影像杂志(2019年1期)2019-04-25
军营文化天地(2018年2期)2018-12-15
中成药(2017年12期)2018-01-19
