
羟亚胺GC/FID定量分析方法的条件优化
2014-08-25 02:44苗翠英徐浩龙
中国人民公安大学学报(自然科学版) 2014年1期
苗翠英, 徐浩龙
(中国人民公安大学刑事科学技术学院,北京 100038)
0 引言
羟亚胺(Hydroxyl imine),分子式: C13H16ClNO,化学名称:1-羟基环戊基-2-氯苯基-N-甲基亚胺基酮,分子量: 237。常见存在形式为盐酸羟亚胺, 高纯度盐酸羟亚胺为乳白色或白色粉末, 熔点175~185 ℃。工业用非高纯度盐酸羟亚胺为咖啡色粉末。羟亚胺和氯胺酮是同分异构体,羟亚胺经过化学反应可得到氯胺酮[1]。作为生产氯胺酮的主要原料,羟亚胺已成为我国新的制毒原料。2008 年4月23 日, 国务院已批准将盐酸羟亚胺列入第一类易制毒化学品。2008年7月8日,公安部、商务部、卫生部、海关总署等6部委联合下发公告,对羟亚胺的生产、经营、购买、运输及进、出口管理进行了规定,以加强对盐酸羟亚胺的严格管制[2-3],进一步有效遏制我国氯胺酮的非法生产及滥用。
羟亚胺和氯胺酮为同分异构体,实际办案中两种物质经常同时检出[4-5]。国内外关于羟亚胺检测方法研究、报道较少。目前国内检验羟亚胺的主要方法为气相色谱/质谱分析方法,该方法可以对羟亚胺进行定性分析[6-7],但不能准确地定量分析。羟亚胺是我国2008年列入管制的易制毒化学品,国际毒品案件中较少出现,目前标准质谱库中没有其质谱图,因此,气相色谱/质谱分析方法检验羟亚胺需人工解析质谱图或通过核磁共振法进行确证[8-9],虽然方法较为准确,但对检验人员专业水平及仪器设备要求较高。为了在实际办案中达到方便快捷的定量效果,关于羟亚胺的定量研究方向有待进一步拓展。
1 实验部分
1.1 主要仪器和试剂
SHIMADZU GC2010 GC/FID(日本岛津公司);
0.01 mg 级天平(瑞典 梅特勒公司);
震荡器(日本 EYELA公司);
移液枪:100 μL~1 000 μL (德国 Eppendorf公司);
瓶口移液器:标称容量10 mL (德国 Brand公司);
进样针:50 μL、100 μL、250 μL (瑞士 Hamilton公司)。
1.2 检测样品
称取盐酸羟亚胺对照品5.77 mg加入甲醇溶液溶解并准确定容至5 mL,制成含盐酸羟亚胺1.154 mg/mL的甲醇溶液。用移液管精密移取上述对照品溶液1 mL,加入甲醇溶液定容至10 mL,制成盐酸羟亚胺浓度为0.115 4 mg/mL(羟亚胺碱浓度为0.1 mg/mL )的样本溶液。
1.3 实验条件
毒品大部分极性较弱,弱极性色谱柱对于弱极性物质具有广谱适用范围,适用于毒品检测。因此色谱柱选择:DB- 5石英玻璃毛细管柱(30 m×0.32 mm×0.25 μm),弱极性色谱柱;
羟亚胺热稳定性好,因此选择:高温检测温度,300 ℃;进样温度 ,280 ℃;柱终温,280 ℃;
载气 :N2,纯度>99.99% ;尾吹:30 mL/min;进样量:1 μL;燃烧气:H2;燃烧气流速:40 mL/min。
1.4 实验内容
GC/FID的定量效果取决于其分离效果,分离效果的直观表现是理论塔板数。理论塔板数受升温速率、初始柱温、柱流速、分流比的影响。因此本文从升温速率、初始柱温度、柱流速、分流比等4项参数进行优化,以羟亚胺响应峰的理论塔板数、保留时间、峰面积、抗干扰情况以及工作曲线为参考标准,对分析方法的参数进行考察。实验中,各组实验参数设定一个为变量,其他恒定。
1.4.1 升温速率实验
在载气流速1 mL/min、分流比20∶1、初始柱温100 ℃的实验条件下,对升温速率进行考察。实验升温速率设定值从5 ℃/min升至20 ℃/min。
1.4.2 初始柱温实验
在载气流速1 mL/min、分流比20∶1、升温速率16 ℃/min的实验条件下,对初始柱温进行考察。实验初始柱温从50 ℃升至200 ℃。
1.4.3 载气流速实验
在分流比20∶1、初始柱温100 ℃、升温速率16 ℃/min的实验条件下对载气流速进行考察。实验载气流速从0.5 mL/min升至2 mL/min。
1.4.4 分流实验
在流速1 mL/min、初始柱温100 ℃、升温速率16 ℃/min的实验条件下对分流比进行考察。实验分流比从5∶1升至50∶1。
1.4.5 抗干扰实验
在流速 1 mL/min、初始柱温100 ℃、升温速率16 ℃/min、分流比20∶1的实验条件下,将羟亚胺样品加入案件中缴获的摇头丸检材,对本实验样本进行分析,并考察羟亚胺和摇头丸中甲基苯丙胺、咖啡因和氯胺酮的分离度。
1.4.6 外标法定量工作曲线实验
称取羟亚胺标准物质样本50 mg,放入50 mL容量瓶中加入甲醇,配制成1 mg/mL的羟亚胺标准溶液,并依次用甲醇稀释至0.1 mg/mL、0.05 mg/mL、0.001 mg/mL、0.005 mg/mL、0.000 1 mg/mL浓度的羟亚胺溶液分别进样,每个浓度的样品平行进样3次,在流速为 1 mL/min、初始柱温100 ℃、升温速率16 ℃/min、分流比 20∶1的色谱条件下进行分析和检测,用峰面积积分值(Y)对浓度(X)进行线性回归,采用0.1 mg/mL、0.05 mg/mL、0.01 mg/mL、0.005 mg/mL、0.001 mg/mL、0.000 5 mg/mL、0.000 1 mg/mL 7个浓度绘制成线性范围为0.000 1 mg/mL~0.1 mg/mL的羟亚胺标准工作曲线。
1.4.7 精密度实验
在流速 1 mL/min、初始柱温100 ℃、升温速率16 ℃/min、分流比 20∶1实验条件下,连续7天每日连续检测10次羟亚胺样本,考察日间精密度和日内精密度,计算出各自的SD和RSD值,考察本方法的精密度参数。
2 结果与讨论
2.1 升温速率
升温速率与理论塔板数线性关系如图1和图2。实验结果表明,升温速率相对于理论塔板数的变化率明显低于初始温度相对于理论塔板数的变化率,说明升温速率对柱效变化的影响小于初始温度。为维持较高柱效,初始温度不宜变化太大,因此本方法宜优先对升温速率进行实验。
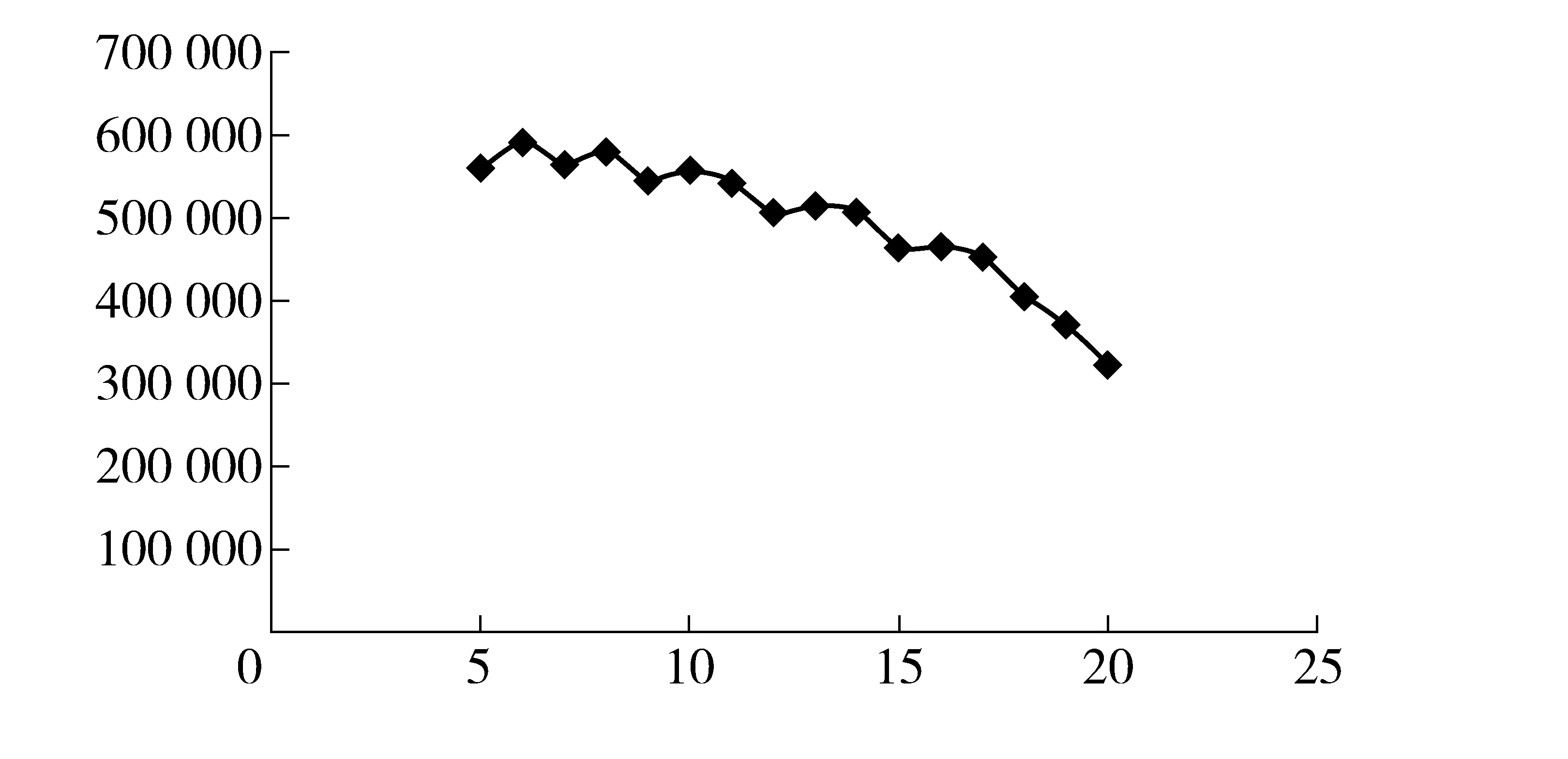
图1 升温速率- 理论塔板数变化曲线图
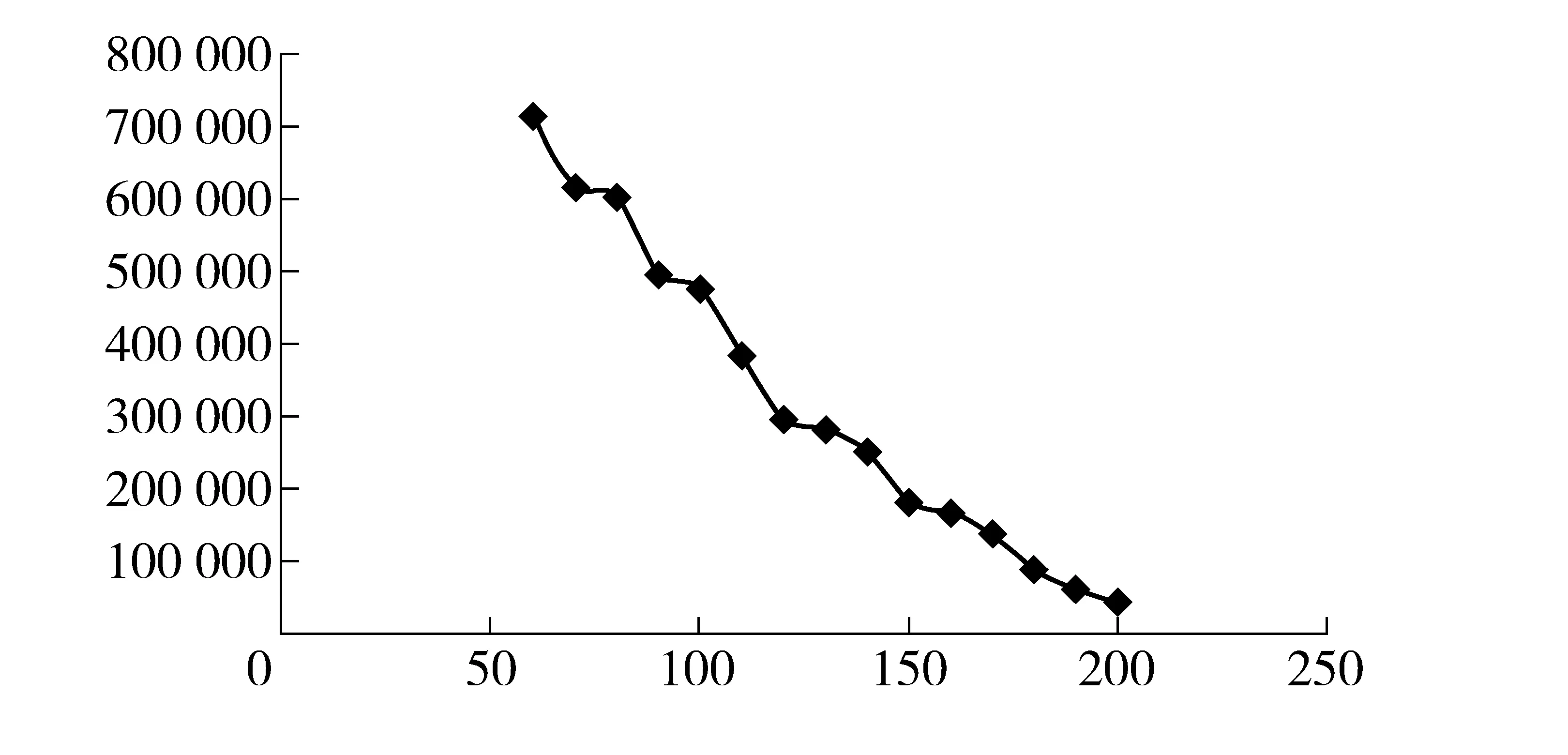
图2 初始温度- 理论塔板数变化曲线图
图1曲线可见,当升温速率在5 ℃/min到20 ℃/min之间变化时,理论塔板数在降低;在16 ℃/min时,理论塔板数下降的斜率增大。为了达到一定的柱效,应从升温速率对柱效变化影响相对较小的稳定区间内选择合适的升温速率,见图3。当升温速率小于或等于16 ℃/min 时,理论塔板数比保留时间的曲线呈上升趋势;当升温速率大于16 ℃/min 时,理论塔板数比保留时间曲线呈下降趋势。
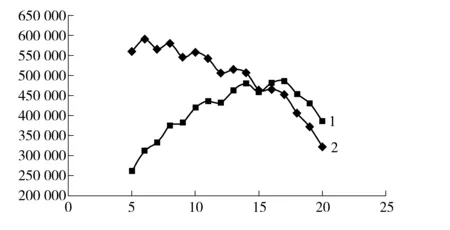
图3 升温速率- 理论塔板数变化曲线图
综上所述,为减少分析时间、提高柱效和保护仪器,以 16 ℃/min为较优升温速率。
2.2 初始柱温
初始柱温与理论塔板数的关系如图4所示。由图 4曲线2可见,初始柱温升高的同时理论塔板数呈线性下降。因进样口温度和初始柱温温差越大,羟亚胺越容易冷凝在色谱柱头,羟亚胺初始色谱带沿柱轴方向展开越小同时分离能力越强。但在实际办案中适用于批量检材处理要求分析时间要适中,本实验拟将时间控制在15 min内。如图4曲线2所示,随着初始温度的增加理论塔板数变化曲线趋势为直线下降,在100 ℃时理论塔板数和保留时间达到最优值。
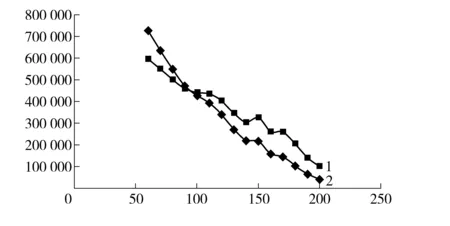
图4 初始柱温- 理论塔板数
综上所述,综合考虑仪器的柱效与待检测组分的保留时间,选择100 ℃作为羟亚胺GC分析方法的初始柱温较为合适。
2.3 载气流速
由图5可见,当柱载气流速在0.5 mL/min~0.8 mL/min 时,理论塔板数较高且理论塔板数随柱流速的升高而显著升高;当流速大于或等于1.1 mL/min 时,理论塔板数开始有较为明显的降低。在0.9 mL/min~1.0 mL/min的范围内理论塔板数较为平稳。
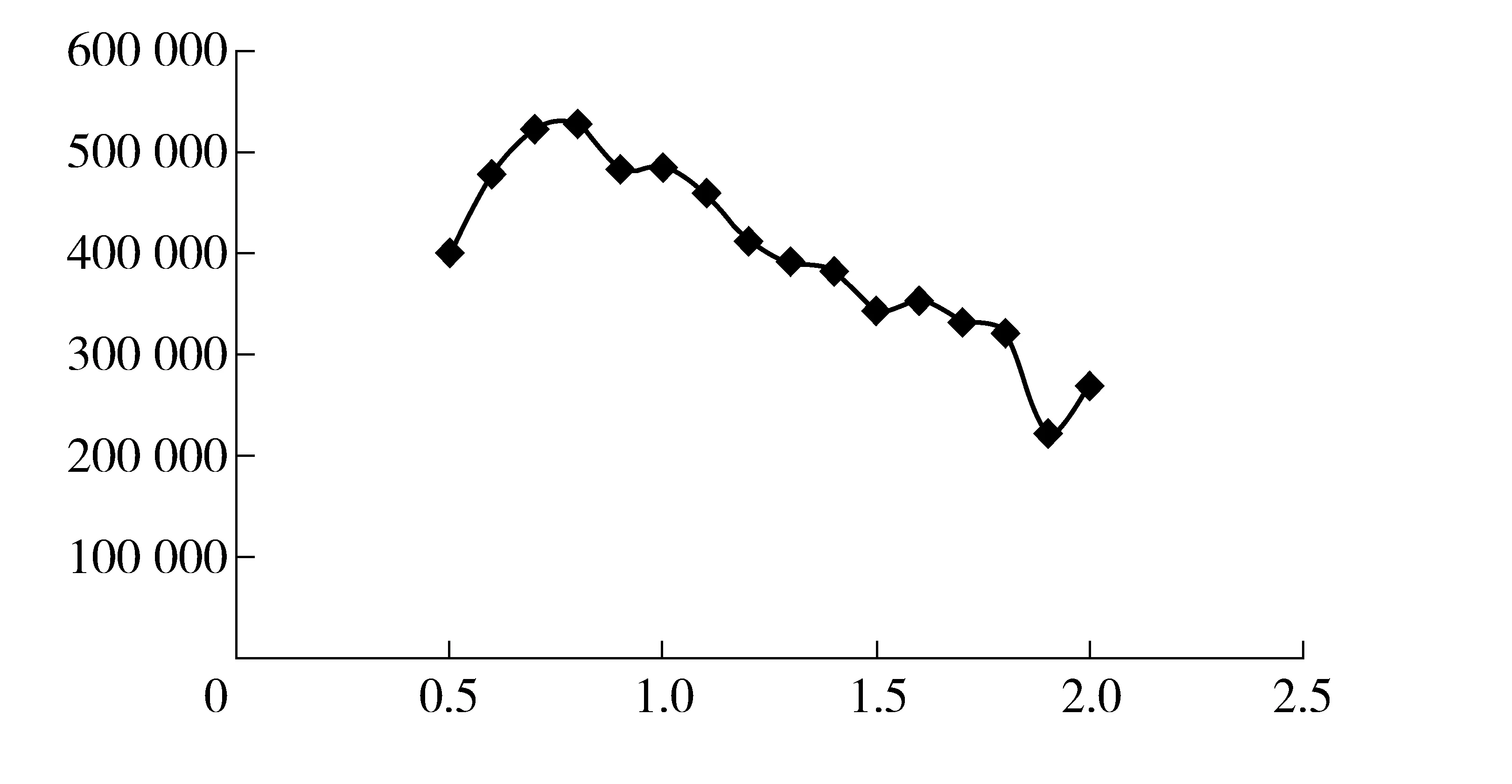
图5 柱流速- 理论塔板数变化曲线图
综上所述,从提高柱效和缩短分析时间两方面综合考虑,选择1 mL/min的柱载气流速较优。
2.4 分流比
分流比将会影响气相色谱的分析结果。在本实验设定的条件下探究分流比与羟亚胺的气相分析检验数据的关系,主要考察羟亚胺的色谱峰面积和理论塔板数。
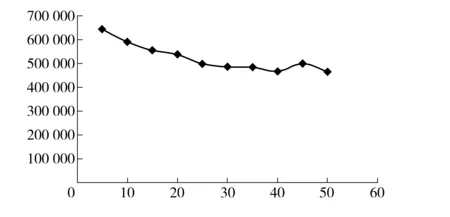
图6 分流比- 理论塔板数变化曲线图
分流比对理论塔板数的影响如图6所示,由图可见分流比对理论塔板数的影响较小。分流比对羟亚胺峰面积的影响如图7所示,其相关曲线拐点为分流比为20∶1,当分流比大于或等于15∶1时,曲线线性程度较高且线性陡峭;当分流比大于或等于20∶1 时,曲线呈一定线性,但线性趋缓且较为平稳。在实际办案中稳定和重现性是重要因素之一,要选择分流比- 峰面积变化曲线较平缓时的分流比,因为在这种参数下分流比有微小的波动不会对峰面积造成较大的影响,较为稳定。同时,如果分流比过大,即实际的进样量会较小,在进样量较小的情况下,检测灵敏度就会降低。
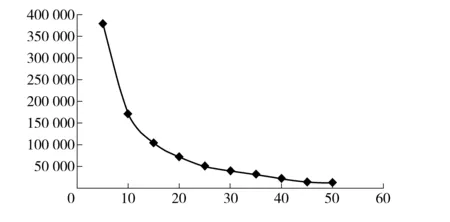
图7 分流比- 峰面积(羟亚胺)变化曲线图
综上所述,20∶1的分流比应用于日常分析检验较为适合。
2.5 抗干扰结果分析
抗干扰分析结果如图8所示,在流速1 mL/min、初始柱温100 ℃、升温速率16 ℃/min、分流比20∶1的实验条件下,羟亚胺和样本摇头丸中甲基苯丙胺、咖啡因和氯胺酮具有较好分离度。羟亚胺和各组分的分离度如下:甲基苯丙胺95.248、咖啡因3.572、氯胺酮14.17。其中咖啡因和羟亚胺的分离度最小,其分离度值为3. 3.572>2,符合实际办案检验要求。
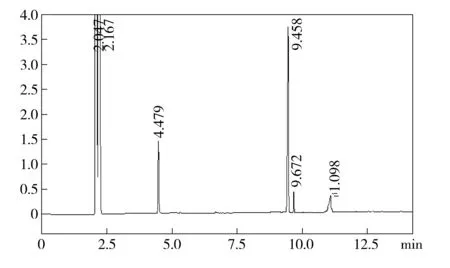
图8 羟亚胺和甲基苯丙胺、咖啡因、氯胺酮色谱峰图
在实际办案中常见的是对同一检材中氯胺酮和羟亚胺的分别定性定量,在本实验优化的羟亚胺GC/FID仪器分析参数下,氯胺酮和各组分的分离度良好,完全满足实际办案中对于羟亚胺的检验要求。
2.6 工作曲线
在本实验所选择的优化实验参数下,GC/FID工作曲线各项参数良好,R值已经达到0.001数量级的要求。 0.000 1 mg/mL~0.1 mg/mL的羟亚胺标准工作曲线如图9所示。
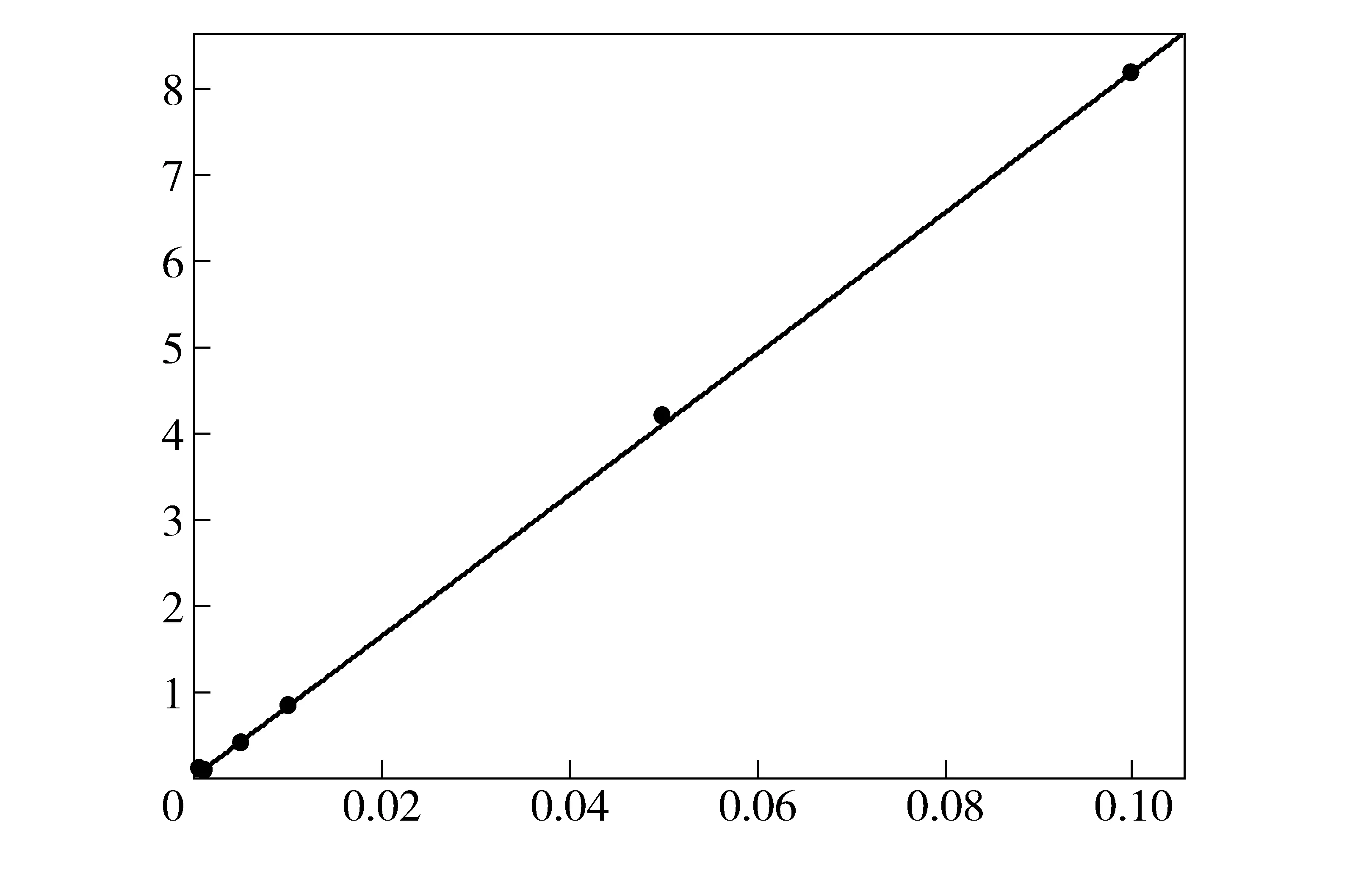
图9 羟亚胺标准工作曲线
0.000 1 mg/mL~0.1 mg/mL标准工作曲线相关技术参数:Y=81 872.7X+23.384 4;R2=0.999 835 1;R=0.999 917 6。
在浓度为0.000 5 mg/mL和0.000 1 mg/mL时羟亚胺的相应理论峰面积应为40和8,但在此时羟亚胺的浓度过小,对仪器要求较高,在此情况下峰面积的计分结果误差较大,本实验的数据明显偏离理论值。因此在本工作曲线所考察的7个浓度点中0.000 5 mg/mL和0.000 1 mg/mL的数据需要排除不计。排除羟亚胺浓度0.000 5 mg/mL和0.000 1 mg/mL后的实验结果表明,本实验所确定的羟亚胺检验鉴定GC/FID仪器的优化实验参数在检测浓度0.001 mg/mL~0.1 mg/mL范围内线性良好,线性回归方程为:
Y=81 999.22X+13.677 89;R2=0.999 836 4;R=0.999 918 2。
在线性回归方程中,本实验所确定的优化实验参数下工作曲线各项参数良好,R值已经达到0.001数量级的要求,符合对毒品或易制毒化学品犯罪案件中检验羟亚胺的定量的要求,定量范围为0.001 mg/mL~0.1 mg/mL。本方法检测限可以达到0.001 mg/mL。
2.7 精密度实验
在优化实验条件下测得的本实验定量精密度数据:
日内精密度为:RSD=3.56(n=10)。日间精密度为:RSD=6.69(n=7)。
3 结论与展望
本文通过对羟亚胺GC/FID实验条件中升温速率、初始柱温、载气流速和分流比4项参数进行考察,得到优化 GC/MS分析条件如下:色谱柱:DB- 5石英玻璃毛细管柱(30 m×0.32 mm×0.25 μm);载气: N2,纯度>99.99%;进样温度:280 ℃;流速:1 mL/min;分流比:20∶1;升温程序:以 100 ℃为初始柱温,以 16 ℃/min 升温至 280 ℃;检测温度:300 ℃;尾吹:30 mL/min;燃烧气:H2,40 mL/min;助燃气:空气,助燃气流速:400 mL/min。
在此分析方法下,羟亚胺浓度为0.001 mg/mL~0.1 mg/mL浓度范围内准确定性,羟亚胺保留时间9.68 min。
该方法能够适用于日常办案需求,定性定量准确、分析速度快。
[1]王世玉,李崇熙. 氯胺酮合成的重排反应改进[J]. 医药工业, 1986(2):49-50.
[2]公安部等六部门. 防止羟亚胺成为制毒原材料[N]. 人民公安报, 2008-07-23(1).
[3]丘燕. 易制毒化学品管理与检测[J]. 广东化工, 2011(5):224-225.
[4]肖翔,吴惠勤,黄晓兰,等.一种制毒原料的气相色谱- 质谱法分析鉴定[J].质谱学报,2009(11):187-189.
[5]高利娜,刘俊亭,杨卓. GC/MS定性K粉及其中间体的应用[J]. 刑事技术, 2008(4): 1558-1617.
[6]钱振华,徐鹏,高利生. GC- MS检验氯胺酮制毒原料羟亚胺1例[J]. 刑事技术,2011(1):65-66.
[7]刘新密,罗国安,张新荣,等.分析化学[M]. 北京:清华大学出版社,2002: 271-255.
[8]肖翔,吴惠勤,黄晓兰,等.一种制毒原料的气相色谱一质谱法分析鉴定[J].质谱学报,2009(11):187-189.
[9]姚汶泉,朱军,刘耀.新型毒品氯胺酮分析方法研究进展[J]. 中国法医学杂志,2007,22(4):253-254.
猜你喜欢
山东医药(2022年19期)2023-01-06
江西通信科技(2022年3期)2022-10-11
预防青少年犯罪研究(2022年1期)2022-08-15
氯碱工业(2022年5期)2022-08-01
安徽化工(2020年5期)2020-10-16
化工管理(2020年19期)2020-07-28
化工技术与开发(2020年6期)2020-06-24
科学技术创新(2020年12期)2020-06-22
电子技术与软件工程(2019年21期)2020-01-16
中国神经精神疾病杂志(2020年9期)2020-01-14
