
乙酰胆碱酯酶与新型香豆素类二聚体抑制剂的作用研究
2014-08-22 06:45王金虎毕海燕
枣庄学院学报 2014年2期
王金虎,毕海燕
(枣庄学院 化学化工与材料科学学院,山东 枣庄 277160)
0 引言
阿尔茨海默氏病(Alzheimer disease, AD)是一种常见的与年龄相关的神经退行性认知功能障碍的疾病,其特征是记忆力减退,神经性精神和行为障碍.AD发病率较高,死亡率仅次于心血管病、癌症及中风而位居第四[1],已成为现代社会严重威胁老年人健康和生命的疾病之一.对症治疗阿尔茨海默氏病的药物主要是一类乙酰胆碱酯酶(AChE)类药物,通过提升中枢胆碱功能,来抑制乙酰胆碱的降解[2].乙酰胆碱(ACh)是一种促进思维活跃的神经递质,AD患者脑内ACh的减少是认知和记忆功能损伤的直接原因.现在市场上治疗AD的药物有五类,其中四个(他克林,多奈哌齐,卡巴拉汀,加兰他敏)是乙酰胆碱酯酶抑制剂(AChEIs)[3-6],而美金刚是N-甲基-D-天冬氨酸受体(NMDA)拮抗剂.但是,所有这些药物在治疗过程中存在副作用,长时间服用药物效力会减弱[7].由于AD病因的复杂多样性,AChEIs等传统单靶向抗AD药物只在一定程度上缓解中、轻度AD患者的病症,并不能终止和逆转AD的病情进展.因此近10多年来新型多靶向抗AD药物的研发日益成为本领域的研究热点.目前对症治疗阿尔茨海默氏病的药物主要属于一类含乙酰胆碱酯酶(AChE)抑制剂的药物.
通过文献阅读在文献[8-9]中收集了十九种表现出结构多样性的香豆素类二聚体化合物作为配体分子(如图1),以考察不同取代基对二聚体类抑制剂与乙酰胆碱酯酶复合构象的影响.本文选用来自老鼠类乙酰胆碱酯酶晶体结构(PDB ID: 1N5M) 作为分子模拟研究中的受体.
1 研究方法
为了得到香豆素二聚体类配体分子的稳定结构,应用Gaussian 03 程序包[10]中密度泛函方法在6-31G(d) 水平下对二聚体类分子1-19进行构型优化,结果如图1所示.优化结束后选取能量低的优势构象用于分子对接研究.应用AutoDock 4.0软件[11]对配体1-19和AChE受体进行分子对接对接.对接过程中,应用ADT中的Ligand 模块设置配体分子的自由度.计算中用来代表受体的grid maps 采用AutoGrid 程序计算.AChE受体分子由于包括两个活性位点,一个为正常的活性区域,另一个为外周阴离子活性区域;为使grid 盒子能够完全包含活性位点及其周围的蛋白区域,本文依次选取格点盒子的大小为60Å×30Å×68Å;每个香豆素二聚体类分子均进行40次独立的对接运算,对接结果根据构象差异按照均方根偏差(RMSD)值等于0.5 Å为标准进行分组,并依据结合自由能的大小对结果进行评估.对每个香豆素二聚体类小分子,选取含有最多对接构象的分组中对接能量最低的构象作为该抑制剂分子的活性构象.
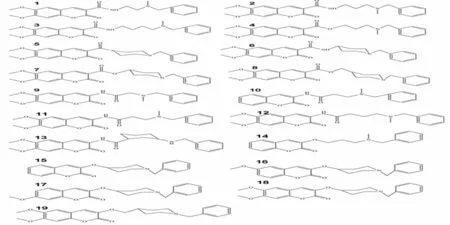
图1 十九种香豆素二聚体类抑制剂分子

图2 十九种香豆素二聚体类小分子优化结构
2 结果和讨论
2.1 分子对接数据图像分析
分组结果表明,在每个香豆素二聚体类分子的40次独立对接结果中,十九种香豆素类二聚体小分子均主要以一种结合方式(构象所占比例≥ 80%)与受体发生相互作用.Autodock 计算的能量项包括分子间作用能(包括范德华相互作用能、氢键相互作用能、去溶剂化能和静电相互作用能),内能和分子扭转能.其中前两项之和为对接能(Docking energy),第一和第三项之和为结合能(Binding energy),对接的能量信息见表1.
正常活性位点由残基Ser203、Glu334和His447组成,外周阴离子活性位点是由His287、Tyr72、Tyr441和Glu292组成.当香豆素类二聚体小分子1、2、3、4、5、6、7、9、11、12、13、18和19在正常活性位点以及二聚体小分子5在外周阴离子活性位点的结合能均为正值,说明这种抑制剂不能与目标蛋白质对接,即使它在电脑模拟计算过程中出现了氢键,结合能仍然为正值,所以在表1中没有出现这几组的数据.
在对接计算的所有能量中,二聚体小分子配体和蛋白酶之间的氢键作用模式最为关键,它直接决定了抑制剂是否能起到抑制作用以及抑制作用的强弱,而配体特定基团的疏水作用则可以很大程度上影响抑制剂的特异性[12-15].分子对接得到的分子与活性口袋及其相邻残基的作用方式见图3和4.通过对接构象与AChE之间氢键作用模式上的差异及图像的研究,可以将对接构象分为三类:(a)以二聚体小分子中6、7号位取代基的部分氧原子与AChE发生氢键相互作用的构象;(b)氢键位于二聚体小分子中的内酯基与AChE的活性口袋残基之间;(c)以二聚体小分子中3号位取代基的部分氧原子、氮原子与AChE发生氢键相互作用的构象.由于不同的香豆素分子可以具有相同的结合模式,因此图3中给出了抑制剂8、14、15和16的在正常活性位点对接构象以作代表;图4给出了抑制剂3、9、11和13在外周阴离子活性位点区域对接的对接构象作为代表.

表1 对接得到的二聚体分子与AChE之间的结合能和对接能

图3 抑制剂8、14、15和16的在正常活性位点区域对接的对接构象
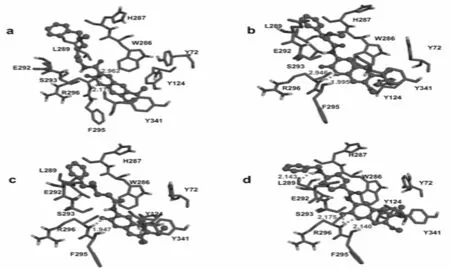
图4 抑制剂3、9、11和13在外周阴离子活性位点区域对接的对接构象
图3a给出了抑制剂8和受体蛋白AChE的正常活性位点相互作用的对接构象.可以看到抑制剂8与活性口袋之间形成了五个氢键相互作用,即抑制剂8的内酯区O13分别与Gly121的HN、Gly122的HN和Ser203的HG形成键长分别为1.941Å、2.170Å和1.758Å的氢键;N20与Try124的OH形成长度为2.445Å的氢键;O8与Glu202的OE1形成长度为2.879 Å的氢键.此构象反映了内酯区氧原子与蛋白质残基形成氢键占主导地位.
图3b显示的是抑制剂14和AChE的正常活性位点相互作用的对接构象.其中抑制剂14的O11与Ser203的HG之间形成了长度为2.089Å的氢键.香豆素类二聚体抑制剂小分子两端的二元环和苯环则位于疏水区域,由于苯环和二元环本身也是疏水的,这种结合模式对抑制剂与目标蛋白的结合有利.图3c呈现了抑制剂15和AChE的正常活性位点相互作用的对接构象.从构象得出抑制剂15的N18与AChE的残基Glu202的OE1之间形成了长度为2.327Å的氢键,形成氢键位置位于蛋白质残基组成的亲水区域,这种构象不利于抑制剂在结合口袋的结合.图3d给出了抑制剂16和AChE的正常活性位点相互作用的对接构象.可以看到抑制剂16与AChE残基之间形成了两个氢键,即抑制剂16的O46分别与Gly122和Ala204的HN形成键长分别为1.728Å和2.075Å的氢键.由于氢键的形成致使抑制剂的疏水的二元环部分位于亲水区,此构象不利于抑制剂的结合.
图4给出的是抑制剂3、9、11和13位于AChE的外周阴离子活性位点时抑制剂和结合蛋白之间的相互作用.和正常活性位点相互作用的对接构象不同的是,这些抑制剂主要利用其内酯区原子和外周阴离子活性位点处残基相互作用,作用形式也主要是氢键相互作用.如抑制剂3的内酯区O13与AChE残基Arg296的O和Arg296的HN之间形成了长度为2.962Å和2.175Å的氢键(图4a);抑制剂9的内酯区O10与AChE残基Arg296的O和Arg296的HN之间形成了长度为2.946Å和1.995Å的氢键(图4b);抑制剂11的内酯区O11与AChE残基Arg296的HN之间形成了长度为1.947Å的氢键(图4c).特别需要指出的是抑制剂13与AChE残基之间形成了三个氢键,即抑制剂13的H45与Leu289的O形成键长为2.143Å的氢键;内酯区O12与O13分别与1N5M残基Arg296的HN之间形成了长度为2.140Å和2.175Å的氢键(图4d).图4a、b、c和d呈现的抑制剂和AChE的外周阴离子活性位点相互作用的对接构象都具有相同的特点,首先氢键的形成大都是由抑制剂内酯区氧原子与结合位点残基作用形成的;其次抑制剂的二聚体环大部分位于残基组成的疏水区,内酯区则位于残基组成的亲水区,这有利于抑制剂与AChE的结合.通过对比研究发现,部分抑制剂有两个负电性中心:抑制剂母体的内脂区和6位或7位的苄基区.由于这两部分负电作用比较强极易与周围蛋白质发生静电作用,从而导致氢键的形成.为了更清楚地认识对接结果中的二聚体类分子在AChE的活性口袋内部取向上的差异,本文对二聚体分子的对接构象进行了叠加,见图5和图6.由图可知,根据二聚体小分子的母体取向可以将其分为三类:抑制剂8和15在正常活性位点区域对接时分子取向相似为第一类,这一类中的化合物与疏水性残基距离较远;抑制剂14和16在正常活性位点区域对接时的所有对接构象母体取向相同,属于第二类,抑制剂3、9、11和13在外周阴离子活性位点区域对接时构象一致和母体取向一致为第三类.
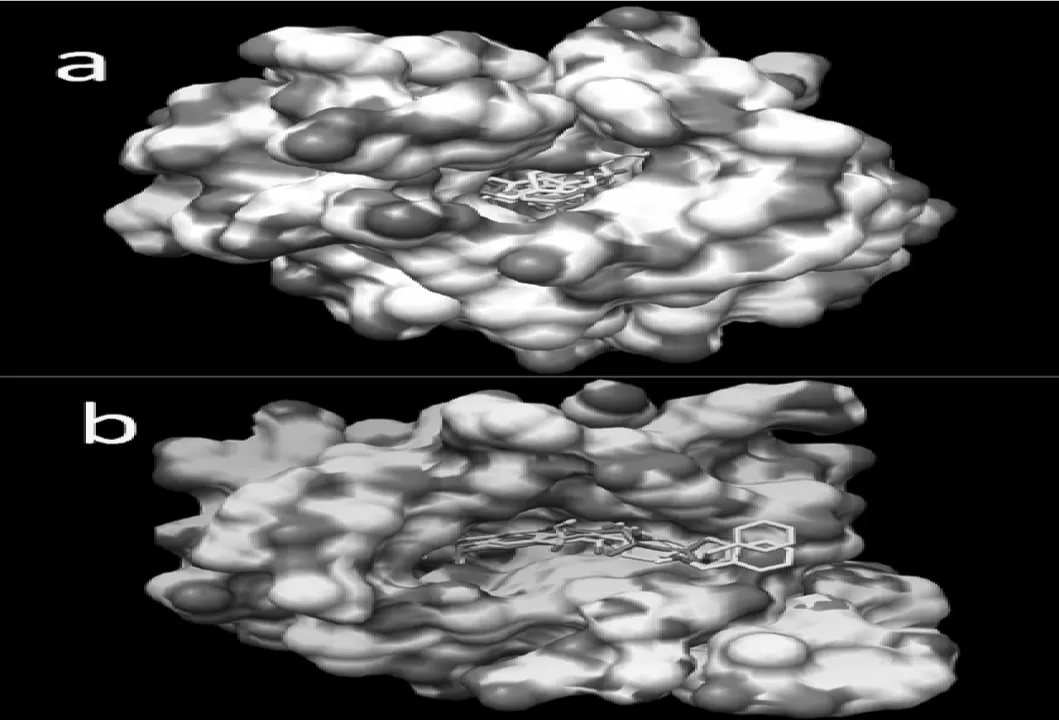
图5 (a)抑制剂8、14、15和16在正常活性位点对接的对接构象在乙酰胆碱酯酶表面的叠加.表面由活性口袋残基周围10 Å范围内的残基构成,白色区域为蛋白质残基组成的疏水区域,其他颜色区域为亲水区域.图中白色、紫色、蓝色和黄色分别代表8、14、15和16抑制剂;(b)抑制剂3、9、11和13在外周阴离子活性位点对接的对接构象在乙酰胆碱酯酶表面的叠加.表面由活性口袋残基周围10Å范围内的残基构成,白色区域为蛋白质残基组成的疏水区域,其他颜色区域为亲水区域.图中白色、紫色、蓝色和黄色分别代表3、9、11和13抑制剂.
研究发现,二聚体类小分子大部分都位于蛋白质残基组成的疏水区域,尤其是小分子两端的二聚体环和苯环几乎都位于疏水区域.但是若小分子6、7号位的取代基有氧原子出现时,该区域有可能位于蛋白质残基组成的亲水区域;由于二聚体小分子母体的内酯基负电作用比较强,极易与周围蛋白质残基发生静电作用产生氢键,所以该位置一般位于蛋白质残基组成的亲水区域;抑制剂3号位取代基上若有氧原子或氮原子出现时,该部位的氧原子或氮原子有可能与周围蛋白质残基发生作用产生氢键,所以该位置有时是位于蛋白质残基组成的亲水区域.
由表1中可以发现,有很多组二聚体小分子由于对接能是正值所以在表中没有出现,尤其是在Ser203、Glu334和His447组成的正常活性位点对接时18组实验中有12组实验的对接能出现正值;而在His287、Tyr72、Tyr441和Glu292组成的外周阴离子活性位点对接时只有1组对接数据出现正值.同时通过对结合能、对接能和抑制常数的比较,说明该香豆素类二聚体抑制剂与乙酰胆碱酯酶作用时,更容易在外周阴离子活性区域与乙酰胆碱酯酶发生作用.在二聚体类分子和乙酰胆碱酯酶相互作用时,它们之间的结合能和对接能与二聚体小分子的抑制作用之间并无良好的线性对应关系,即对接能最低的配体并不是抑制作用最强的,反之亦然.之所以出现这种现象,主要有两个方面的原因: (1)在结合位点相同时,不同香豆素类二聚体小分子的取代基在原子类型、原子数目以及自由度等方面差别较大,这是导致结合能和对接能差异较大的一个关键因素;(2)决定抑制剂抑制效果的因素有很多,除了结合能以外,受体和配体之间的轨道相互作用、极化作用、疏水性相互作用也是影响抑制剂活性的关键因素.
3 结论
研究结果表明,部分抑制剂有两个负电性中心:抑制剂母体的内酯区和6位或7位的苄基区.由于这两部分负电作用比较强极易与周围蛋白质发生静电作用,从而导致氢键的形成.香豆素二聚体类小分子主要以内酯区与AChE结合.这使其有可能成为一种有更好应用前景的新型乙酰胆碱酯酶抑制剂.
[1]段金海,汪华侨,陈少琼. 阿尔茨海默病患者脑白质损害与认知功能的关系[J].中华神经科杂志,2006,39(2):76- 79.
[2]Bartus R T, Dean R L, Beer B, Lippa A S. A Review of the Behavioral Effects of AF64A, A Cholinergic Neurotoxin .[J] Science, 1982,217: 408-417.
[3]Whitehouse, P. J. [J] Acta Neurol. Scand. Suppl. 1993, 149: 42-45.
[4]Kelly, C. A.; Harvey, R. J.; Cayton, H. [J] BMJ 1997, 314: 693-694.
[5]Scott, L. J.; Goa, K. L. [J] Drugs 2000, 60, 1095-1122.
[6]Gottwald, M. D.; Rozanski, R. I. [J] Expert Opin. Investig. Drugs 1999, 8, 1673-1682.
[7]Thies W, Bleiler L. Alzheimer's Association. Alzheimer's Association report: 2012 Alzheimer's disease facts and figures. [J] Alzheimers Dement, 2012, 8: 131-168.
[8]Marco C, Leonardo P, Francesco L, Orazio N, Paolo P, Angela S,Saverio C, Angelo C. Design, synthesis and biological evaluation of coumarin alkylamines as potent and selective dual binding site inhibitors of acetylcholinesterase. [J] Bio Med Chem, 2013, 21: 146-152.
[9]Yue qing H, Jun Z, Oormila C, Fanny C F, Nancy Y. Design, synthesis and evaluation of novel heterodimers of donepezil and huperzine fragments as acetylcholinesterase inhibitors. [J] Bio Med Chem, 2012, 20: 5678-5698.
[10]Gaussian 03 , Revision A.3. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A.; Vreven, Jr., T. ; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V. ; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; and Pople, J. A. Gaussian, Inc., Pittsburgh PA, 2003.
[11]Goodsell D, Morris G M, Olson A J. Automated docking of flexibleligands: applications of AutoDock. [J] Mol Recognit,1996,9:1-5.
[12]De Winter H L, von Itzstein M. Aldose reductase as a target for drug design: molecular modeling calculations on the binding of acyclic sugar substrates to the enzyme. [J] Biochem, 1995, 34(26): 8299-8308.
[13]Hess B, Bekker H, Berendsen H J C, Fraaije J G E M. LINCS: A linear constraint solver for molecular simulations. [J] Comp Chem, 1997, 18(12): 1463-1472.
[14]Srinivasan N, Golbeck J H. Protein-cofactor interactions in bioenergetic complexes: the role of the A1A and A1B phylloqui nones in Photosystem I. [J] Biochim Biophys Acta, 2009, 1787(9): 1057-1088.
[15]Hunger H D, Schmidt G, Flachmeier C, Behrendt G, Coutelle C. High-sensitivity protein detection by a new "contact-copy" method using a protein A-neomycin phosphotran sferase II fusion protein. [J] Anal Biochem, 1990, 186(1): 159-164.
猜你喜欢
同位素(2022年6期)2022-12-30
物理学报(2022年10期)2022-06-04
唐山师范学院学报(2020年6期)2020-04-16
农药科学与管理(2019年8期)2019-11-23
唐山师范学院学报(2019年3期)2019-06-18
天然产物研究与开发(2018年10期)2018-11-06
天然产物研究与开发(2018年8期)2018-09-10
天然产物研究与开发(2018年6期)2018-07-09
天然产物研究与开发(2018年4期)2018-05-07
天然产物研究与开发(2018年2期)2018-04-04
