
1,1-二氟乙烷气相脱氟化氢制备氟乙烯催化剂研究进展
2014-08-21 09:05王术成林胜达于万金刘武灿
化工生产与技术 2014年6期
王术成 张 迪 林胜达 于万金 刘武灿
(浙江省化工研究院有限公司,国家ODS替代品工程技术研究中心,杭州310023)
氟乙烯(VF)的化学式为CH2=CHF,在常温下为无色,有微弱醚味的气体,相对分子质量为46.04,常压下沸点为-72.3℃。VF是一种重要的含氟单体,可用于生产聚氟乙烯(PVF),具有耐候性、良好的机械强度、化学惰性、低透气性和透水性等优点。PVF应用广泛,因而它的合成广受关注[1-2]。
目前,工业制备VF主要有2条途径,1种是通过乙炔(C2H2)和氟化氢(HF)反应制得[3-7]:
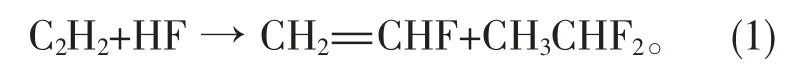
但是这种直接合成VF的方法存在收率低和催化剂寿命短等缺点,且使用易燃易爆的乙炔作为原料非常危险。
另1种是通过1,1-二氟乙烷(R152a)脱HF制得[8-9]。这种制备VF的方法收率高且产品纯度高,杜邦公司已采用该工艺大规模生产VF。此外,该生产装置简单且反应过程易于控制。R152a脱HF制备VF反应为平衡反应,而且VF的收率和反应温度密切相关,反应式如下:
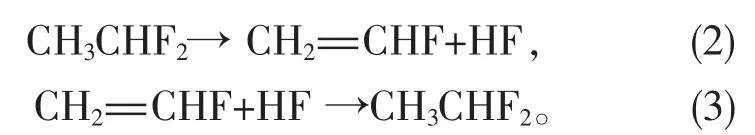
R152a气相脱HF是最有前途制备VF的方法之一,但是脱HF催化剂在实际应用中存在易失活的问题,而稳定性是催化剂实际应用最重要的方面之一,因此开发高活性和良好抗积碳的催化剂是该技术的关键。目前为止,仅有少量关于该催化剂体系的研究。本文综述了脱HF催化剂的研究进展。
1 脱HF反应机理
氢氟烷烃脱HF反应生成氢氟烯烃是按照E1消去机理(碳正离子机理)进行,属于典型的吸热反应3CH[10-11]。
Li等报道,CF3CH3脱HF反应的活性中心是L酸性位,该反应按照碳正离子机理分2步进行。首先,电负性的氟原子和催化剂的酸性位之间因强相互作用而使得C—F键异裂,并且是决速步骤,然后活泼的+CF2CH3碳正离子中间体轻易脱去H+生成CF2=CH2[12]。
Teinz等报道,AlF3脱HF反应的高活性源于其强酸性,CF3CH3中的F原子和催化剂中的强酸中心的相互作用引发脱HF反应。C—F键的断裂是决速步骤,因此L酸性决定了脱HF反应的活性[13]。
2 R152a气相脱HF制备VF
气相催化脱HF制备VF的催化剂包括铬基催化剂(Cr2O3/CrF3)和铝基催化剂(Al2O3/AlF3)等。但是,铬基和铝基催化剂酸性强,而在强酸上易形成积碳和/或聚合物[12,14]。催化剂表面形成积碳是反应过程中催化剂严重失活的原因,为了抑制积碳,添加合适的助剂调变酸性有望解决这一问题。如在Cr2O3/CrF3或Al2O3/AlF3催化剂中添加过渡金属氧化物和(或)碱土金属氧化物能调变催化剂的表面酸性。此外,文献报道的反应中,稀土金属氧化物能有效降低积碳和提高催化剂稳定性[15-18]。上述的金属氧化物可使L酸中心中毒,随着含量的增加,强酸的酸量随之降低。通过添加助剂减少Cr2O3/CrF3或Al2O3/AlF3催化剂的表面强酸,从而抑制积碳,提高催化剂的稳定性[6]。
早在20世纪50年代,Harmon公开了通过R152a脱HF制备VF的方法,所述脱HF催化剂包括氧气、活性碳和IA、IB、IIA、IIB、VB和VIII族的金属、氧化物或盐类化合物等,可使用的盐类包括硼酸盐、溴化物、氯化物、铬酸盐和铬铁矿等。如使用氟化钙催化剂,500℃时,R152a转化率为66%。但反应需在高温条件下进行否则转化率相对较低[8]。
开发更高效的R152a脱HF催化剂得到持续关注,近些年,研究重点是具有更高活性和稳定性的铬基和铝基催化剂。
Christoph等报道,优选的铬基催化剂是结构主要为α-Cr2O3的催化剂(尤其是基本为α-Cr2O3的催化剂),可通过热分解(NH4)2Cr2O7或400~500℃加热处理无定形的Cr2O324~48h制得[19-21]。另一个方法是将如翠铬绿(Guignet′sgreen)的含B2O3铬催化剂在氧气或空气中,在较高温度下加热处理,然后在200~400℃条件下用HF处理以除去B2O3。如使用通过热分解(NH4)2Cr2O7制得的α-Cr2O3催化剂,在温度350℃、接触时间12s条件下,产物中VF、R152a和C2H2的质量分数分别为74.7%、21.5%和2.2%。相较于比表面积为200m2/g的无定形铬催化剂,α-Cr2O3的转化率和稳定性都更好。因此使用α-Cr2O3而非无定形铬,能够提高铬基催化剂的活性和稳定性。
Rao等报道,使用一种通过共沉淀法制备的结晶钴取代的α-Cr2O3的含铬催化剂组合物,进一步改进制备VF的方法[22]。将氮气和R152a通入到使用质量分数分别为95%的铬和5%钴催化剂的反应器中,氮气和R152a摩尔比为4:1、接触时间为15s,在250℃时,反应产物VF和R152a质量分数分别为36.4%和63.1%;在350℃时,反应产物VF和R152a质量分数分别为82.0%和16.7%。表明结晶钴取代的α-Cr2O3催化剂有更好的活性。
以上的铬基催化剂具有良好的活性,但是C2H2等副产会增加分离纯化VF的成本。为了降低副产C2H2的含量和提高催化剂寿命,降低反应温度是有效的方法之一,但同时会降低时空收率。相比之下,铝基催化剂显示更好的选择性和稳定性。
Nappa等公开了裂解催化剂包括至少1种选自镁、锌及其混合物的氧化物、氟化物和氟氧化物中的化合物和任选至少1种选自铝的氧化物、氟化物和氟氧化物中的化合物,其中,除镁和锌之外的任何金属原子(如铝)和所述催化剂中镁和锌的总原子比例约为1:4,或者更低。含有1种或者多种金属氟化物的金属氟化物催化剂制备方法如下:将金属卤化物或硝酸盐溶于去离子水配成水溶液,加入质量分数48%的氢氟酸搅拌处理,持续搅拌过夜,然后将浆料用蒸气浴干燥,得到干燥的固体在400℃焙烧得到。如在275℃和接触时间0.32s条件下,使用MgF2/AlF3(1:9)和β-AlF3催化剂上,R152a转化率分别为27%和12%。在250~280℃和足够的接触时间条件下,报道的催化剂至少能得到R152a脱HF制备VF转化率平衡值的80%。相比之下,根据已报道的基于VF和R152a摩尔比得到的VF平衡浓度,过去的方法在227℃时得到质量分数13%的VF,327℃时得到质量分数40%的VF[23]。
Rao等报道脱HF催化剂包括MgF2/β-AlF3、ZnF2/β-AlF3和MgF2-ZnF2/β-AlF3,其中Mg与Al、Zn与Al、Mg+Zn、Al的摩尔比都为1:1。通过加热单一相的氟化组合物MAlF5(H2O)2或NH4MAlF6(H2O),其中M是至少1种选自锌和镁的2价金属[24-25]。使用5mLZnF2/β-AlF3催化剂,在275℃和接触时间6s条件下,反应1h和720.4h,R152a的转化率分别为34.3%和35.3%,催化剂稳定性良好。300℃条件下,MgF2/β-AlF3催化剂反应1h和48h后,R152a的转化率分别为42.8%和43.4%,且VF选择性都为100%。该方法副产C2H2含量低且催化剂寿命长。
目前,性能优良的铬基和铝基催化剂得到很多关注,此类催化剂的R152a转化率和VF选择性都很高。通过添加合适的助剂调变酸性得到中等酸强度的改性催化剂兼具高活性和稳定性。
3 其他脱HF催化剂的应用
氢氟烷烃通过气相脱HF这一途径制备包含VF在内的氢氟烯烃近年来得到广泛研究,其他脱HF催化剂和R152a气相脱HF催化剂在制备方法和性质方面有很多相似之处,因此有必要研究其他脱HF催化剂的应用。
1,1,1-三氟乙烷(R143a)脱HF制备偏氟乙烯(VDF),1,1,1,2-四氟乙烷(R134a)脱HF制备1,1,2-三氟乙烯(R1123),1,1,1,2,3-五氟丙烷(R245eb)或1,1,1,2,2-五氟丙烷(R245cb)脱HF制备2,3,3,3-四氟丙烯(R1234yf),1,1,1,3,3-五氟丙烷(R245fa)脱HF制备1,3,3,3-四氟丙烯(R1234ze)1,1,1,2,3,3-六氟丙烷(R236ea)或1,1,1,2,2,3-六氟丙烷(R236cb)脱HF制 备1,2,3,3,3-五 氟 丙 烯(R1225ye),1,1,1,3,3,3-六氟丙烷(R236fa)脱HF制备1,1,3,3,3-五氟丙烯(R1225zc)的催化剂包括添加过渡金属(如Fe、Zn、Ni、Co、Mn、Zr、Cu等)氧化物,碱土金属(如Mg、Ca等)氧化物,稀土金属(如La、Y、Ce等)氧化物的铬基和铝基催化剂[26-37]。
总之,氢氟烷烃在合适的催化剂作用下可通过气相脱HF高效制得氢氟烯烃,通过添加合适的助剂调变酸性制备中等酸强度的脱HF催化剂是今后的研究重点。
4 结论与展望
R152a气相脱HF是最有前途制备VF的方法之一,该方法收率高且产品纯度高。
R152a脱HF反应的活性中心是L酸,L酸性决定了脱HF反应的活性,但是催化剂强酸位上形成积碳是催化剂严重失活的原因。气相脱HF催化剂包括具有强酸性的铬基和铝基催化剂。通过添加合适的助剂调变酸性制备中等酸强度的脱HF催化剂是其保持高活性和稳定性的关键。
为了抑制积碳,在催化剂中添加过渡金属氧化物,碱土金属氧化物和稀土金属氧化物降低强酸的酸量有望解决这一问题。制备高活性和良好抗积碳,用于脱HF反应的催化剂是今后的研究重点,开发适用于氢氟烷烃气相脱HF制备不同氢氟烯烃的催化剂是今后的发展方向。
[1]Scheirs J.Fluoropolymers:Technology,Markets and Trends[R].Shawbury:Rapra Technology Ltd,2001.
[2]Ebnesajjad S.Polyvinyl Fluoride:Technology and Applications ofPVF[R].PresidentFluoro ConsultantsGroup,LLC,Elesevier,2013.
[3]Petit R,Kaziz C,Wetroff G.Process for hydro-fluorination of acetylenic hydrocarbons:US,3187060[P].1965-06-01.
[4]Christoph FJ,Elkton JR.Process for preparing vinyl fluoride and 1,1-difluoroethane:US,3395187[P].1968-07-30.
[5]Dupont.Process for preparing vinylfluoride and 1,1-difluoroethane:GB,1026105[P].1966-11-08.
[6]BiQY,Qian L,Xing LQ,etal.Vaporphase hydro-fluorination of acetylene to vinylfluoride over La2O3-Al2O3catalysts[J].Journal of Fluorine Chemistry,2009,130(6):528-533.
[7]BiQY,Lu JQ,Xing LQ,etal.Effect ofcalcinationtemperature on La-Modified Al2O3catalysts for vapor phase hydrofluorination of acetylene to vinylfluoride[J].Chinese Journal of Chemical Physics,2010,23(1):89-94.
[8]Jesse H.Preparation of Vinyl Fluoride:US,2599631[P].1952-06-10.
[9]Benjamin FS.Dehydrofluorination of1,1-difluoroethane:US,2674632[P].1954-04-06.
[10]Tavoularis G,Keane M A.Gas phase catalytic dehydrochlorination and hydrodechlorination ofaliphatic and aromatic systems[J].JournalofMolecularCatalysis A:Chemical,1999,142(2):187-199.
[11]Kamiguchi S,Watanabe M,Kondo K,et al.CatalyticdehydrohalogenationofalkylhalidesbyNb,Mo,Ta,andhalideclusterswithanoctahedralmetalframeworkandbyarechloride cluster with a triangularmetal framework[J].Journal o fMolercular Catalysis A:Chemical,2003,203(1/2):153-163.
[12]LiG L,NishiguchiH,Ishihara T,etal.Catalytic dehydrofluorination of CF3CH3(HFC-143a)into CF2CH2(HFC-1132a)[J].Applied Catalysis B:Environmental,1998,16(4):309-317.
[13]Teinz K,Wuttke S,Börno F,et al.Highly selectivemetal fluoride catalysts for the dehydrohalo-genation of 3-chloro-1,1,1,3-tetrafluorobutane[J].Journalof Catalysis,2011,282(1):175-182.
[14]Nickkho A M,Eltananny G,Wuttke S,etal.A comparative study of surface acidity in the amorphous,high surface area solids,aluminium fluoride,magnesium fluoride(III)or aluminium(III)fluorides[J].Journal of Fluorine Chemistry,2008,129(5):366-375.
[15]Parmaliana A,Arena F,Frusteri F,et al.Magnesiasupported Nickel catalysis II.Surface properties and reactivity inmethane steam reforming[J].JournalofCatalysis,1993,141:34-47.
[16]Martínez R,Romero E,Guimon C,et al.CO2reforming of methane over coprecipitated Ni-Al catalystsmodified with lanthanum[J].Applied Catalysis A:Genernal,2004,274:139-149.
[17]Trimm D L.Catalysts for the control of coking during steam reforming[J].Catalysis Today,1999,49(1/3):3-10.
[18]Liu B S,Au C T.Carbon deposition and catalyst stability over La2NiO4/γ-Al2O3during CO2reforming ofmethane to syngas[J].Applied Catalysis Genernal,2003,244:181-195.
[19]Frank J C,George W C,Mallikarjuna R.Treatment of chromium oxide and catalytic manufacture of vinylfluoride:US,6359183[P].2002-03-19.
[20]GumprechtW H,Manzer L E,Rao V N M.Process for the manufacture of 1,1,1-trifluoro-dichloroethane and 1,1,1,2-tetrafluoro-chloro-ethane:US,4843181[P].1989-06-27.
[21]Lerou JJ.Chromium oxide catalystcomposition:US,5036036[P].1991-07-30.
[22]Rao V NM,Rosenfeld H D,Sievert A C.Cobalt-substituted chromium oxide compositions,their preparation,and their use as catalysts and catalyst precursors:US,7217678[P].2007-05-15.
[23]NappaM J,Rao VNM.Catalyticmanufactureofvinylfluoride:US,6262321[P].2001-07-17.
[24]Rao V N M,Munirpallam A S Catalytic manufacture of vinylfluoride:US,5880315[P].1999-03-09.
[25]Rao V N M,Subramanian M A.Catalysts for halogenated hydrocarbon processing,theirprecursorsand theirpreparation and use:US,5559509[P].1996-09-24.
[26]Younosuke A,HeikichiS.Preparation of vinylidene fluoride:JP,54130507[P].1979-10-09.
[27]Maher Y E.Process for converting a 1,1,1-tri-fluoroalkaneInto a 1,1-difluoroalkene:EP,0234002[P].1987-09-02.
[28]PowellR L,SharrattAP.Process for the production of fluorine containing olefins:US,5856593[P].1999-01-05.
[29]Serge H,Schlrlmann L A.Process for the preparation of trifluoroethylene:FR,2710054[P].1995-03-24.
[30]Serge H.Dehydrofluorination of fluoroalkane into fluoroalkene:FR,2729136[P].1996-07-12.
[31]Nappa M J,Rao U N M,Sievert A C.Tetra-fluoropropene production processes:WO,2008002500[P].008-01-03.
[32]Yoshikawa S,Kaneda S.Production of 1,3,3,3-tetrfluoro propene:JP,11140002[P].1999-05-25.
[33]Michael V D P.Fluorinated propenes from penta-fluoropropane:US,5986151[P].1999-11-16.
[34]Wang H,Tung H S.Process for the production of HFO trans-1234ze from HFC-245fa:EP,2014637[P].2009-01-14.
[35]Rao V N M,Subramanian M A.Fluoroolefin manufacturing process:US,6031141[P].2000-02-29.
[36]Hirokazu A,Eiji S.Method for manufacturing 1,1,1,2,3-pentafluoropropene 1,1,1,2,3-penta-fluoropropane:US,5679875[P].1997-10-21.
[37]Vanderpuy M,Cook G R,Scheidle PH,et al.Process for themanufacture of fluorinated olefins:WO,2008057794[P].2008-05-15.
猜你喜欢
天然产物研究与开发(2019年1期)2019-03-01
汽车观察(2018年10期)2018-11-06
科学与技术(2018年8期)2018-04-26
——会偷偷侵蚀你的发动机!
人民交通(2016年8期)2017-01-05
浙江大学学报(工学版)(2016年11期)2016-06-05
合成技术及应用(2015年2期)2016-01-10
柴油机设计与制造(2015年3期)2015-12-05
电源技术(2015年11期)2015-08-22
装备环境工程(2015年4期)2015-02-28
化工科技(2014年4期)2014-06-09
