
在体大鼠心肌缺血再灌注损伤中NF-κBp65与AngⅡ的关系
2014-08-14 11:27王芬珍
重庆医学 2014年15期
杨 锐,王芬珍
(1.浙江省宁波市第一医院心血管内科 315000;2.福建省福州市心血管病研究所 350009)
随着20世纪80年代心脏介入治疗学的推广,心肌缺血再灌注损伤(IRI)成为一大研究热点。其中NF-κB及AngⅡ均参与了心肌IRI的发生发展,在此病理过程中两者的关系也备受关注。已有研究表明,NF-κB是AngⅡ的前体血管紧张素原再合成的重要调节因子[1],NF-κB激活可增加组织AngⅠ及AngⅡ水平,并在转录水平影响AT1RmRNA表达[2];同时AngⅡ也能激活NF-κB,且有研究认为AngⅡ诱导促炎细胞因子表达是通过NF-κB/IκB信号转导通路实现的。在此,我们将进一步探讨心肌IRI中NF-κB与AngⅡ的关系。
在前期实验的基础上,我们观察到缺血30min再灌注60 min(I30minR60min)为 NF-κBp65蛋白表达的高峰点,利用抗氧化剂四氧吡咯二硫氨基(PDTC)及咪达普利分别作为NF-κB和血管紧张素转化酶(ACE)的特异性抑制剂,探讨NF-κBp65与AngⅡ在心肌IRI中的关系及其机制。
1 材料与方法
1.1 实验动物及分组 雄性健康清洁型SD大鼠32只,体质量250~300g,由福建医科大学动物中心提供。许可证号SCXK(闽)2004-0002,参照文献报道的方法稍加改进建立动物模型[3]。按照随机数字表法分为4组:(1)IR组:缺血30min再灌注60min;(2)PDTC+IR组:PDTC 125mg/kg,用0.85%的生理盐水稀释,于缺血前15min腹腔注射;(3)咪达普利+IR组:咪达普利3mg/kg,用0.85%的生理盐水稀释,于缺血前15min颈外静脉缓慢推注;(4)PDTC+咪达普利+IR组:PDTC 125mg/kg及咪达普利3mg/kg分别于缺血前15min腹腔注射及颈外静脉缓慢推注。
1.2 标本的采集与处理 实验结束相应时间点取材。(1)取血:含蛋白酶抑制剂的真空试管备用,迅速下腔静脉取血2mL加入试管,用于血浆AngⅡ水平的检测;(2)取组织:迅速剪取平行房室沟下2mm处的左室游离壁心肌,分成2块,一块在液氮中速冻后保存到-80℃冰箱用于核蛋白提取;另一块新鲜组织尽快匀浆用于组织AngⅡ水平的检测。
1.3 免疫组织化学分析法 定位和定量检测NF-κBp65活性:光学显微镜下观察心肌细胞,如出现棕黄色颗粒为阳性表达,活化的 NF-κBp65定位于细胞核;在高倍显微镜下(×200),每组切片选取10个视野,细胞总数不少于500个,计算阳性细胞数的百分数,用阳性表达率表示NF-κBp65活化蛋白的核易位表达量。
1.4 ELISA法检测心肌组织NF-κBp65蛋白转录活性 提取心肌组织细胞核蛋白,按照考马斯亮蓝蛋白测定试剂盒说明书,定量细胞核蛋白数量,按TransAM NF-κBp65蛋白活性检测试剂盒说明书进行ELISA。
1.5 放射免疫分析法测定血浆和心肌组织AngⅡ含量 按试剂盒说明书,采用均相竞争放射免疫分析法直接测定血浆和心肌中的AngⅡ含量。
1.6 统计学处理 采用SPSS13.0统计软件进行分析。计量资料以表示,各组间比较采用两因素两水平的析因分析,以P<0.05为差异有统计学意义。
2 结 果
2.1 各组NF-κBp65蛋白核易位变化及NF-κBp65蛋白转录活性比较 由图1、表1可见:PDTC+IR组、咪达普利+IR组及PDTC+咪达普利+IR与IR组相比,均显著抑制NF-κBp65蛋白核易位变化,两药联合效果更明显,各组之间差异均有统计学意义(均P<0.05);进一步检测 NF-κBp65DNA转录活性,同样,各药物预处理组与IR组相比也可显著抑制NF-κBp65蛋白的DNA转录活性,两药联合效果更明显,各组之间差异均有统计学意义(均P<0.05)。

图1 免疫组织化学分析法测定各组NF-κBp65蛋白核易位变化(×400)
表1 药物预处理对NF-κBp65蛋白核易位的影响(,n=8,%)

表1 药物预处理对NF-κBp65蛋白核易位的影响(,n=8,%)
*:P<0.05,与IR组比较。
核阳性表达率IR组组别53.82±6.22 PDTC+IR组 30.37±3.44*咪达普利+IR组 43.20±4.58*PDTC+咪达普利+IR组 12.26±1.03*
2.2 各组血浆及心肌组织AngⅡ水平的变化 由表2可见,各药物处理组与IR组相比,血浆及组织AngⅡ水平显著下降,两药联合效果更明显,各组之间差异均有统计学意义(均P<0.05)。
表2 药物预处理对血浆及组织AngⅡ水平的影响(,n=8)
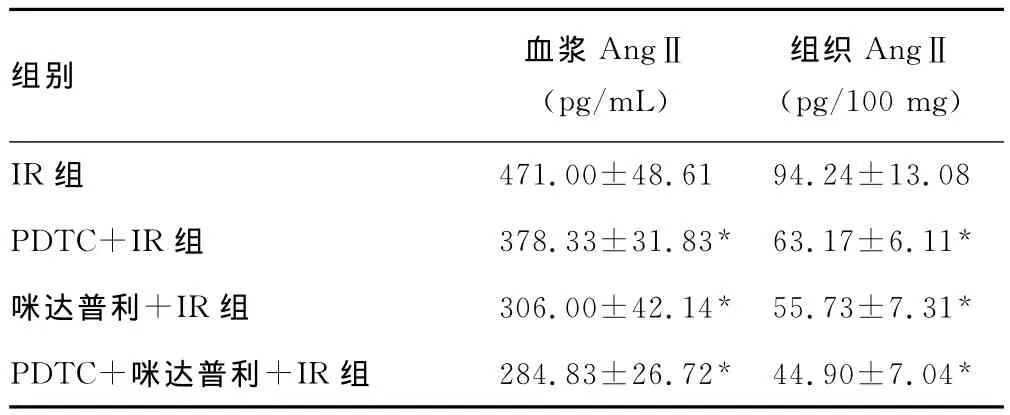
表2 药物预处理对血浆及组织AngⅡ水平的影响(,n=8)
*:P<0.05,与IR组比较。
组别 血浆AngⅡ(pg/mL)组织AngⅡ(pg/100mg)IR组471.00±48.6194.24±13.08 PDTC+IR组 378.33±31.83* 63.17±6.11*咪达普利+IR组 306.00±42.14* 55.73±7.31*PDTC+咪达普利+IR组 284.83±26.72* 44.90±7.04*
3 讨 论
PDTC是NF-κB的特异性抑制剂,主要作用是:(1)具有抗氧化性,体外研究证实它可直接清除反应性氧中介物或与其相关酶的金属离子发生螯合作用,抑制酶活性[4],从而抑制NF-κB激活;(2)通过阻止IκB与 NF-κB分离、阻碍 NF-κB核易位等途径阻断NF-κB激活的信号转导通路[5];(3)具有促氧化作用,通过螯合Cu2+产生的复合物能氧化NF-κB的巯基而抑制其活性[6]。
血管紧张素转化酶抑制剂(ACEI)中的咪达普利是新一代、长效ACEI类药物,其活性代谢产物咪达普利拉与体内ACE结合,降低ACE活性,高选择性作用于肾素血管紧张素醛固酮系统(RAS),从而强效而持久的抑制AngⅡ的生成[7]。
从实验中笔者不难发现,与IR组比较咪达普利预处理在显著降低血浆及组织AngⅡ含量的同时也伴随着NF-κBp65蛋白活性的下降,提示咪达普利可能通过抑制AngⅡ的表达在一定程度上抑制NF-κBp65活性。已有多项研究表明ACEI和/或AngⅡ受体阻滞剂可通过抑制NF-κB激活对心肌IRI的病理过程有所改善[8-9]。AngⅡ可以作为一个内源性促炎症因子激活 NF-κB从而增加氧化应激等作用[10]。Dostal等[11]指出AngⅡ可作为一个生长调节因子,促进多种细胞生长因子的合成和释放包括 NF-κB。Muller等[12]报道转肾素、血管紧张素原双基因大鼠心肌细胞、血管内皮细胞和平滑肌细胞NF-κBp65蛋白表达增加,提示AngⅡ介导的心血管损害与NF-κBp65活性有关。Marta等[13]发现AngⅡ可提高培养的大鼠血管平滑肌细胞中NF-κB的结合活性,AngⅡ受体阻滞剂可阻断这一作用并呈剂量依赖性。因此笔者推测在心肌IRI中,激活的AngⅡ甚至RAS可激活NF-κB转录系统,两者共同参与此病理过程。
其次笔者发现,与IR组比较PDTC预处理在显著抑制NF-κBp65蛋白DNA转录活性的同时也伴随血浆及组织AngⅡ含量的降低,提示PDTC可通过抑制NF-κBp65的激活在一定程度上抑制AngⅡ。Muller等[12]曾指出,PDTC可通过抑制NF-κB激活保护AngⅡ诱导的炎性反应及器官损伤。Brasier等[14]发现,NF-κB是 AngⅡ前体-血管紧张素原再合成的重要调节因子,因此AngⅡ的致炎作用在一定程度上依赖NF-κB的调节。对AT1受体基因序列分析也发现其启动序列含有NF-κB的结合序列,提示NF-κB可能会诱导AT1受体表达[15]。因此,笔者推测在心肌IRI中,NF-κBp65转录系统的激活对AngⅡ甚或RAS的激活有影响,两者共同参与此病理过程。
最后,从药物预处理的角度上,笔者发现咪达普利、PDTC或两药联合预处理均抑制AngⅡ与NF-κBp65活性,ACEI类药物对心脏的保护已毋庸置疑,PDTC的使用仍处于实验室阶段,以此为新的靶点,可能为临床防治心肌IRI提供更多的途径。
综上所述,心肌IRI是多种生物活性因子参与的复杂病理过程,涉及多种复杂细胞信号转导机制,其中AngⅡ或RAS与NF-κBp65可相互激活,并在多层次和诸多环节存在交互作用。
[1]Brasier AR,Jamaluddin M,Han Y,et al.AngiotensinⅡinduces gene transcription through cell-type-dependent effects on the nuclear factor-kappaB (NF-κB)transcription factor[J].Mol Cell Biochem,2000,212:155-169.
[2]Cowling RT,Zhang X,Reese VC,et al.Effect of cytokine treatment on type 1AangiotensinⅡreceptor(AT1A)transcription and splicing in rat cardiac fibroblasts[J].Am J Physiol Heart Circ Physiol,2005,289(3):H1176-1183.
[3]刘付平,姚宏伟,李俊.大鼠心肌缺血再灌注损伤模型的改进[J].安徽医科大学学报,2003,38(3):234-236.
[4]Boyle EM Jr,Kovacich JC,Canty TG Jr,et al.Inhibition of nuclear factor-kappa B nuclear localization reduces human E-selectin expression and the systemic inflammatory response[J].Circulation,1998,98(19):282-288.
[5]Lakshminarayanan V,Lewallen M,Frangogiannis NG,et al.Reactive oxygen intermediates induce monocyte chemotactic protein-1in vascular endothelium after brief ischemia[J].Am J Pathol,2001,159(4):1301-1311.
[6]Kis A,Yellon DM,Baxter GF.Second window of protection following myocardial preconditioning:an essential role for PI3kinase and p70S6kinase[J].J Mol Cell Cardiol,2003,35(9):1063-1071.
[7]王吉云,胡大一.较高选择性ACE抑制剂-咪达普利[J].中国医药导刊,2000,2(3):39-41.
[8]Das UN.Angiotensin-Ⅱ behaves as an endogenous pro-inflammatory molecule[J].J Assoc Physicians India,2005,53:472-476.
[9]Bond M,Chase AJ,Baker AH,et al.Inhibition of transcription factor NF-kappaB reduces matrix metalloproteinase-1,-3,and -9production by vascular smooth musele cells[J].Cardiovasc Rse,2001,50(3):556-565.
[10]Das UN.Angiotensin-Ⅱ behaves as an endogenous proinflammatory molecule[J].J Assoc Physicians India,2005,53:472-476.
[11]Dostal DE,Hunt RA,Kule CE,et al.Molecular mechanisms of angiotensinⅡin modulating cardiac function:intracardiac effects and signal transduction pathways[J].J Mol Cell Cardiol,1997,29(11):2893-2902.
[12]Muller DN,Dechend R,Mervaala EM,et al.NF-kappaB inhibition ameliorates angiotensinⅡ-induced inflammatory damage in rats[J].Hypertension,2000,35(2):193-201.
[13]Marta Ruiz-Ortega,Oscar Lorenzo,Monica Ruperez,et al.AngiotensinⅡactivates nuclear transcription factorκB through AT1and AT2in vascular smooth muscle cells[J].Circ Res,2000,86:1266-1272.
[14]Brasier AR,Recinos A 3rd,Eledrisi MS.Vascular inflammation and the renin-angiotensin system[J].Arterioscler Thromb Vasc Biol,2002,22(8):1257-1266.
[15]Cowling RT,Gurantz D,Peng J,et al.Transcription factor NF-kappa B is necessary for up-regulation of type 1 angiotensinⅡreceptor mRNA in rat cardiac fibroblasts treated with tumor necrosis factor-alpha or interleukin-1 beta[J].J Biol Chem,2002,277(8):5719-5724.
猜你喜欢
中国卫生标准管理(2022年21期)2023-01-03
温州大学学报(自然科学版)(2022年2期)2022-05-30
天然产物研究与开发(2018年7期)2018-08-21
制导与引信(2017年3期)2017-11-02
上海农业学报(2017年3期)2017-04-10
工业设计(2016年11期)2016-04-16
医学研究杂志(2015年2期)2015-06-10
中国当代医药(2015年16期)2015-03-01
中国药理学通报(2014年2期)2014-05-09
俄罗斯问题研究(2012年1期)2012-03-25
