
药厂生产环境的监测及风险评估
2014-08-10 10:04:52徐华
化工与医药工程 2014年5期
徐华
(上海朗脉洁净技术股份有限公司,上海 200100)
国内外多年的实践与研究结果证明,造成药品发生微生物污染等质量问题,其中一个重要原因是生产环境不符合要求,而制药车间长期稳定的空气洁净是保证药品质量的关键环节之一。
随着季节、天气等大环境的变化,外界空气中污染生物微粒和非生物尘埃微粒、温湿度和压差等对制药车间空气洁净和药品质量可能会造成影响,所以为保证制药车间能满足生产要求,进行环境的连续监控是非常必要的。
1 基本概念
1.1 BMS(Building Management System)
从广义上讲,BMS 即“楼宇管理系统”,它将建筑物或建筑群内的变配电、照明、电梯、空调、供热、给排水、消防、保安等众多分散设备的运行、安全状况、能源使用状况及节能等,实行集中监视、分散管理和控制的建筑物管理与控制系统;
从狭义上讲,在制药工程中,大家普遍认可的BMS 含义及范围,主要以空调系统为主,实现对区域内洁净度、温湿度、压差梯度等的控制;简单而言,即根据生产环境的需求,在PLC/DCS 系统及现场仪表、阀门等硬件设备支持下,编写相应控制程序,实现控制功能并达到设计目标。
1.2 EMS(Environment Monitoring System)
EMS 即环境监测系统,目的是通过对药品生产环境的监测,评估是否符合要求,是对BMS 的控制结果进行验证,同时评估生产环境的变化趋势,为用户的风险管理提供必要信息;后面章节我们将对其内容和范围做详细论述。
需要注意的是,工程设计中通常将BMS 和EMS划分为两个独立系统,目的是简化空调净化控制系统的 CSV 复杂性。对于制药行业的自动化系统,目前遵循或参考的GAMP5(Good Automation Manufactory Practice/良好自动化生产实践指南-第五版)是由国际制药工程协会(ISPE)主编的实践指南,虽然它不属于法规,但却是目前国际制药行业进行计算机化系统验证方法的主要参考依据, 同时也是医药自动化最重要的合规性指南。
参考GAMP5 相关章节的描述,自动化系统的软件可分为四类[1]:
- 第1 类软件:基础结构软件(Infrastructure Software)
- 第3 类软件:不可配置软件(Non-Configured Products)
- 第4 类软件:可配置软件(Configured Products)
- 第5 类软件:定制应用软件(Custom Application)
对于BMS 的编程,为了满足控制需求,需要有针对性地开发定制功能块,归类为第5 类软件,这就需要做源代码,即“白盒”验证。
而EMS 的目的是监视数据,可直接调用符合国际电工委员会IEC 61131-3 制定的可编程逻辑控制器标准功能块,完成数据采集、处理工作,属于第4 类软件,只需要做功能性测试,即“黑盒”验证。
如EMS 与BMS 共用一套系统,虽然减少了硬件投资,但因整体控制程序属于第5 类软件,势必大大增加验证的工作量和复杂性;所以两者独立,是目前空调净化自动化系统应对验证合规性检查的最有效方式。
与BMS 不同,EMS 是对最终结果的监测和评估,也是GMP 认证中的重点——相对“过程”而言,更注重“结果”;下面我们详细阐述如何设计EMS,并通过风险评估,使系统符合GMP 规范要求。
2 环境监控参数与范围
遵循GMP(2010 版)[2]关于药品生产区域的环境参数(一般规定):
- 药品生产区域的环境参数主要包括空气洁净度(尘粒数和微生物数)、温度和湿度、新鲜空气量、压差、照度、噪声等。
- 为了保证药品生产质量、防止生产环境对药品的污染,生产区域必须满足规定的环境参数标准。
- 药品生产区域应以空气洁净度(尘粒数和微生物数)为主要控制对象,同时还应对其温度、湿度、新鲜空气量、压差、照度、噪声等参数作出必要的规定,其中至少应对温度、湿度、压差、悬浮粒子、微生物进行验证。
所以在制药行业,根据生产环境的关键参数要求,通常EMS 监控范围可从以下四方面考虑。
2.1 一粒三微的监视
即悬浮粒子、浮游菌、沉降菌、表面微生物。
2.2 环境温度和相对湿度的监视
洁净室(区)的温度和相对湿度应与其生产及工艺要求相适应(无特殊要求时,温度在18~26 ℃,相对湿度在45%~65%为宜)[2]。
2.3 微压差的监视
不同净化级别区域的压差不小于10Pa[2],我们一般控制在 10~30Pa 的范围内,超过30Pa 时将出现开门困难,50~70Pa 时,门/窗缝隙将出现啸叫。
2.4 微风速的监视
A 区是高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。单向流系统在其工作区域必须均匀送风,风速为0.36~0.54 m/s(指导值,通常我们控制在0.45m/s±20%),应当有数据证明单向流的状态并经过验证[2]。
3 环境监测方式的选择
对于环境参数的监测可分为在线式和离线式,以主要控制对象悬浮粒子为例,所用仪器为激光粒子传感器。
3.1 优缺点比较
离线式优缺点比较见表1。

表1 离线式优缺点
在线式优缺点比较见表2。
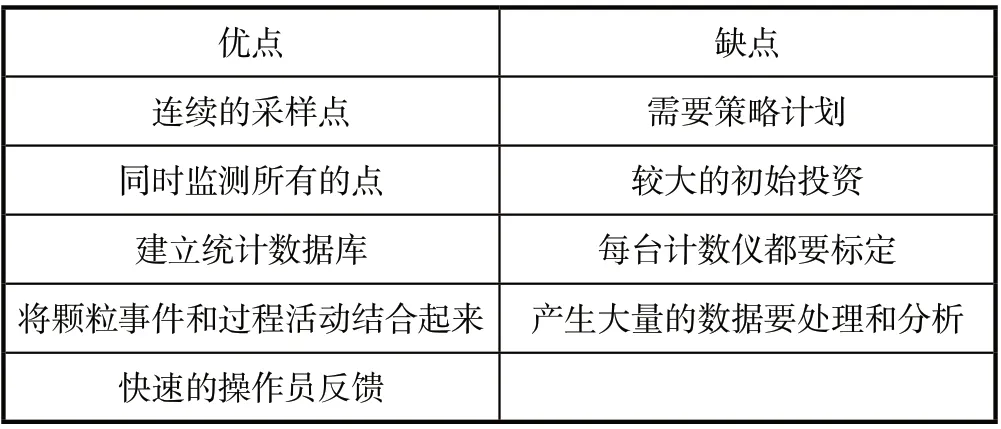
表2 在线式优缺点
3.2 相关法规
3.2.1 GMP(2010 版)相关规定[2]
第十条应当按以下要求对洁净区的悬浮粒子进行动态监测:
(1)根据洁净度级别和空气净化系统确认的结果及风险评估,确定取样点的位置并进行日常动态监控。
(2)在关键操作的全过程中,包括设备组装操作,应当对A 级洁净区进行悬浮粒子监测。A 级洁净区监测的频率及取样量,应能及时发现所有人为干预、偶发事件及任何系统的损坏。
第十一条应当对微生物进行动态监测,评估无菌生产的微生物状况。监测方法有沉降菌法、定量空气浮游菌采样法和表面取样法(如:棉签擦拭法和接触碟法)等。动态取样应当避免对洁净区造成不良影响。成品批记录的审核应当包括环境监测的结果。
3.2.2 欧盟相关规定[5]
Grade A: Continuous monitoring required during setup and operation.
(A 级区:在设备的装配和操作全过程连续监测。)
3.2.3 FDA 相关规定[6]
Critical areas: Continuous monitoring during setup and operation.
(关键区域:在设备的装配和操作全过程连续监测。)
3.3 二种检测方式的应用分析
综合上面的描述,可以发现离线监测存在以下不可克服的缺点:
- 对洁净厂房带来额外的人员和设备,增加了洁净负荷;
- 缺乏采样点和采样时间的固定性;
- 所取得的数据在生成报告时缺乏一致性和连续性;
- 检测报告是滞后的结果;
- 耗费大量的人力资源。
在生产过程中,环境的情况往往是变化的,由于离线监测无法提供连续监测数据,因此无法估计系统是在何时偏离了规定工况,更无法估计产品的质量。
在线监测系统避免了离线监测的种种缺陷;粒子、浮游菌、温湿度、压差和风速等相关传感器安装在各个点,将连续测量结果传送到计算机,通过统一的监视平台,全面实时反映生产环境的状态。
所以在线连续监测符合相关法规和实际需求,可以及时反馈环境参数是否超标,通过预警值提示操作人员;完备的历史数据记录及分析功能,为环境变化趋势及风险评估提供可靠依据,而其缺点也可通过优化设计予以克服。
4 环境监测点的选择
通过前面的描述,在线环境监测系统的优化设计,其中最重要的一个环节是如何布置监测点。监测点并非越多越好,增加费用的同时,也导致大量的数据需要处理,缺乏分析重点。
我们的原则是:符合相关法规和生产需求的前提下,尽量简化,节约投资。
4.1 环境监测布点的设计
下面以一条冻干生产线的粒子检测布点为例。
4.1.1 环境监测布点的规范设计
按照ISO14644-1 或GB 医药工业洁净室(区)悬浮粒子的测试方法[3,4]所提出的检测布点见图1。
采样点数量:
4.1.2 环境监测布点的实际设计
图2 显示了冻干车间检测布点的实际设计。
“7”号位为B 级背景,选择靠近门,除B 背景区的监测外,可兼顾人流进出的风险;B 级区的监测点通常通过风险评估,只要安装一个就可以符合要求。
如果产品还没被完全封口,考虑装填区在冻干机之前是A 级区,所以“5” 和 “6”号位需要监测。

图1 遵循规范的冻干线粒子布点

图2 实际生产的冻干线粒子布点
4.2 环境监测的布点原则
从以上两个设计中,我们可以得出,选择粒子监测点,参考ISO 14644-1 等的要求,如采取洁净区面积开根号取整数原则,无疑这个数量是比较大的,而且没有真正监测到风险点;在线粒子监测的设计中,除了参考规范外,还要考虑药厂的实际生产情况,基于产品性质、工艺特点、操作属性、风险评估结果来确定。
4.2.1 GMP(2010 版)中规定[2]
根据洁净度级别和空气净化系统确认的结果及风险评估,确定取样点的位置并进行日常动态监控。
4.2.2 欧盟相关规定[5]
Monitoring locations based on a formal risk analysis study and the results obtained during classification.
(监测位置的选择应该基于正式的分析评估研究以及在验证过程中获得数据。)
4.2.3 FDA 相关规定[6]
Measurements taken at sites where there is the most potential to the exposed sterilized product, containers, and closures.
(测量点选择在风险最大的位置,如暴露的无菌药品、容器和瓶塞。)
4.3 环境监测点选择的分析
综合以上法规中相关描述,我们采取的原则是:
空气直接和药品接触的区域一定布点,空气可能和药品接触的区域按背景区的布点规则布点,空气不和药品接触的区域不需要布点 。
其他温湿度、压差、风速等参数的布点原则与之类似;不同工艺及产品有差异,需要做针对性的优化设计,这里限于篇幅,不做详细论述,概括几条:
- 选择能够最具备代表性的环境参数;- 方便取样,取样操作不会对环境产生新的污染 ;- 监测点不会受到其他干扰,对监测结果产生影响。
5 生产环境的风险评估
前面阐述了关于环境监测系统(EMS)的基本设计概念,归根结底,我们需要通过EMS 提高或保证药品生产环境,尽量规避风险的产生或发生后如何处理。
风险一直存在于我们的药品生产过程中,但如何对风险进行量化?下面通过风险分析理论,做一简要说明。
5.1 划分风险等级
我们从风险发生的可能性和严重性两方面分析,量化得出风险等级。
5.1.1 风险发生的可能性
对于风险发生可能性的估计(基于连续的操作过程),这些可能性有以下几个等级:
低(Low)= 该风险每年出现一次;
中(Medium)= 该风险每月出现一次;
高(High)= 该风险每周出现一次。
5.1.2 风险造成的严重性
对风险造成结果的严重性估计:
低(Low) = 轻微的后果,其造成的影响会快速消退;
中(Medium) = 中等的后果,其造成影响处于短期与中期之间;
高(High) = 严重的后果,会造成长期影响或短期的灾难性影响。
图3 是关于可能性与严重性的综合评估,得出初步的风险等级(Class1~3):
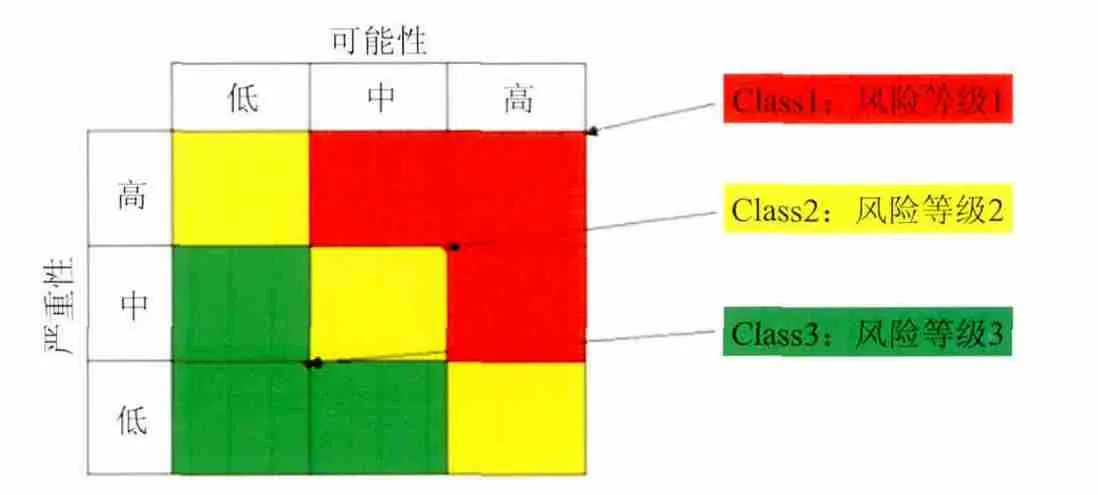
图3 风险等级模型
5.2 划分优先等级
风险等级确定后,我们下一步需要对于这些风险的可监测性做一评估和量化;对于风险情境,估计其被发现的可能性有以下几个等级:
低(Low) = 发生三次的情况下,可能只有一次被发现,或者更少,因而可能性较低,相应的风险度就高;
中(Medium) = 发生两次的情况下,可能一次被发现,因而可能性中等;
高(High) = 很可能每次发生都发现,因而可能性高,由于侦测性高,风险性就低。
综合风险等级和可监测性,根据图4 模型,一个风险情境的优先等级展现出来,从中可以判断哪个情境需要给予措施。
5.3 风险评价及应采取的措施
用来最小化风险情境及其影响程度的措施种类:低(Low) = 对于低优先等级的,没有必要进行一般的日常测试,而是建议进行一些安装测试;
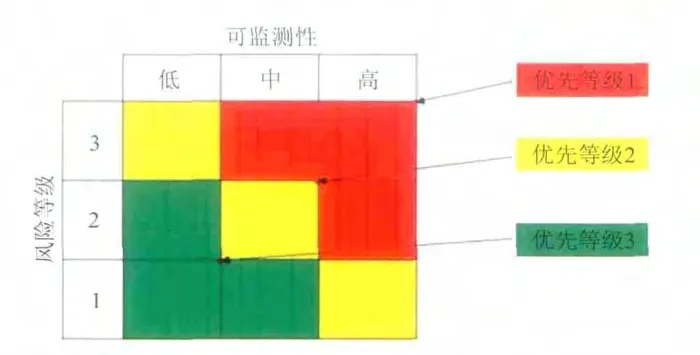
图4 优先等级模型
中(Medium) = 对于中等优先等级的,建议进行安装测试,而一般的日常测试应该在验证总结报告中给予描述和评价;
高(High) = 高优先等级意味着高风险,因而需要更多的测试来最小化风险,这些测试可能会包含系统的改变,应该进行日常的测试。
根据以上理论,以某灌装生产线为例,得出表3的风险评价。

表3 某灌装生产线风险评价表(样表)
6 结束语
EMS 的设计,首先需要熟悉GMP 等相关法规,其次针对不同药品生产需求,运用风险控制理论进行深化设计,选择合适的产品和方案,最终完成一套性价比高的环境监测系统。
[1] ISPE Headquarters (Corporate Author), GAMP5-A Risk-based Approach to Compliant GxP Computerized Systems[Z]. April 1, 2008. ISBN-13: 978-1931879613.
[2] 国家卫生部.药品生产质量管理规范(2010 年修订)[S].
[3] EU, Annex 1-Manufacture of Sterile Medicinal Products[Z]. March 1, 2009.
[4] ISO14644-1,洁净室和相关受控环境-空气洁净度等级划分[S].
[5] GBT16292-2010,医药工业洁净室(区)悬浮粒子的测试方法[S].
[6] FDA, Guidance for Industry-Sterile Drug Products Produced by Aseptic Processing – CGMP[S]. September 2004.
猜你喜欢
电子制作(2019年19期)2019-11-23 08:41:54
资源节约与环保(2019年7期)2019-01-22 07:42:50
石油石化绿色低碳(2019年6期)2019-01-14 01:16:20
中国资源综合利用(2017年4期)2018-01-22 02:46:51
湖南城市学院学报(自然科学版)(2016年2期)2016-12-01 04:07:06
公民与法治(2016年4期)2016-05-17 04:09:15
中国资源综合利用(2016年12期)2016-01-22 02:02:27
中国交通信息化(2015年12期)2015-06-06 06:53:33
应用数学与计算数学学报(2014年1期)2014-09-26 12:19:03
电测与仪表(2014年18期)2014-04-04 12:33:08
