
希特林蛋白缺乏症的研究进展
2014-08-10 12:28堵向楠丁岩综述王向波审校
疑难病杂志 2014年9期
堵向楠,丁岩综述 王向波审校
综 述
希特林蛋白缺乏症的研究进展
堵向楠,丁岩综述 王向波审校
希特林蛋白缺乏症;SLC25A13;新生儿肝内胆汁淤积症;成年发作II型瓜氨酸血症
希特林蛋白缺乏症(citrin deficiency)是一类常染色体隐形遗传性疾病。最初见于日本,由Kobayashi等[1]于1999年通过对18例患者的外周血标本进行分析,发现了致病基因SLC25A13,该基因编码的蛋白称为希特林蛋白。SLC25A13基因突变可以在某个环节上影响其肝内酶的活性,并继发性的降低该酶的水平,最终影响尿素循环导致氨代谢障碍,但并不直接影响编码精氨酸琥珀酸合成酶(argininosuccinate synthetase,ASS)的基因本身的结构,机制至今仍不清楚。精氨酸琥珀酸合成酶是尿素循环中的限速酶,它的缺乏可导致瓜氨酸蓄积,精氨酸生成减少及高氨血症,引发类似于肝性脑病的表现。由于成人发病的希特林蛋白缺乏症比较少见,且由于其发病时易与精神科疾病相混淆,导致临床误诊,因此我们就希特林蛋白缺乏症目前的研究现状进行综述。
1 流行病学
Kobayashi等[2]报道的SLC25A13突变基因的携带率在中国(包括台湾)为1.54%,日本为1.46%及韩国0.90%,提示Citrin缺乏症在东亚广泛存在;同时他们发现在中国SLC25A13基因的携带者以长江为界存在显著的南北差异。研究表明,NICCD的发病率为1/19 000,而CTLN2发病率为1/230 000~1/100 000[3,4]。
2 病因与发病机制
SLC25A13编码的Citrin蛋白全长675个氨基酸,相对分子质量为74kDa,与SLC25A12基因编码的Aralar蛋白在氨基酸序列上具有77.8%的同源性。但Citrin蛋白主要在肝脏表达,而Aralar则主要在肌肉表达,两者分布不同。编码Citrin蛋白的mRNA主要在肝脏、肾脏、心肌及成纤维细胞中均有表达,产生Citrin蛋白。但是以肝脏细胞内表达为主,提示Citrin蛋白缺乏症是一种局限于肝脏的疾病[5]。故SLC25A13基因突变致肝脏内Citrin表达减少时可因肝脏内精氨酸生成减少而使肾脏等其他组织细胞代偿性的增加Citrin蛋白的表达,因此CTLN2型患者血液中的精氨酸可以维持在正常或略升高的水平。
Citrin蛋白位于肝细胞线粒体内膜,是一种钙结合性跨膜溶质载体蛋白,负责将线粒体中的天冬氨酸转运至胞浆,同时将细胞质的谷氨酸转运至线粒体内部。这一过程与苹果酸穿梭相偶联,同时将细胞质中还原型烟酰胺腺嘌呤二核苷酸(NADH)被重新氧化为NAD+,从而维持了细胞质中氧化/还原状态的稳定性。
Citrin蛋白的缺乏可导致NADH大量堆积,进而使乳酸等还原性物质的糖异生过程受阻,引起低血糖和高乳酸血症等生化异常,还可激活枸橼酸/苹果酸穿梭系统,使枸橼酸在转运出线粒体后在细胞质中被其裂解酶分解产生大量乙酰辅酶,进而合成脂肪酸和脂肪,过多的脂肪堆积可以引起高脂血症及肝脂肪变性等表现,同时可抑制半乳糖代谢酶UDP-葡萄糖-4-表位酶的活性,导致半乳糖血症,严重者可出现白内障等表现。可见,细胞质中NADH的堆积是本病发生发展的关键环节。由于大量碳水化合物可加重胞浆中NADH的堆积,故过量摄入碳水化合物通常是导致本病急性发作或者加重病情进展的诱因[6]。此外,Citrin蛋白缺乏时,细胞质中的天冬氨酸不足,这种氨基酸作为合成蛋白质的原料在缺乏时则会影响蛋白质的合成,而且通过与瓜氨酸合成精氨酰琥珀酸而参与尿素合成,因此出现低蛋白血症、高氨血症和瓜氨酸血症的临床表现[7~9]。
3 临床特点
2001年,Abukawa等[10]与Tomomasa等[11]分别发现希特林蛋白缺乏还可在新生儿或婴幼儿时期引起特殊类型的肝炎及黄疸的发生。由于它们的临床表现并不相同,临床上分为两类:第一类为希特林蛋白缺乏所致新生儿肝内胆汁淤积症(neonatal intrahepatic cholestasis caused by citrin deficiency,NICCD),第二类为成年发作II型瓜氨酸血症(adult-onset type II citrullinemia, CTLN2)。
3.1 新生儿肝内胆汁淤积 Ohura等[12]于1997年首先报道1例出现一过性的瓜氨酸血症的新生儿肝炎病例,并于2001年发现该类患者在SLC25A13基因上具有与CTLN2相同变异。Tomamasa等[11]于2002年将这种在新生儿及婴幼儿人群中出现的特殊类型的肝炎,同时伴有多种氨基酸血症、胆汁淤积、低蛋白血症、半乳糖血症和低血糖为主要表现的疾病,命名为NICCD并阐述如下:本病发病年龄多在2个月龄以内,很少晚于5个月龄,男女比例无差异,平均出生体质量低于正常。主要临床表现有:(1)新生儿或婴儿期起病,有肝大、黄疸等婴儿肝炎综合征表现,部分患儿可有凝血功能障碍,可有白内障等半乳糖血症表现;(2)发育迟缓;(3)血生化检测可发现胆红素(直接胆红素为主)、胆汁酸、酶学指标(如GGT,ALP,AST,ALT等)等升高,而白蛋白/总蛋白降低,同时有不同程度高血氨、高乳酸血症,往往伴甲胎蛋白明显增高;(4)血氨基酸分析发现瓜氨酸、苏氨酸、蛋氨酸、酪氨酸和精氨酸增高;(5)尿液分析可有半乳糖、半乳糖醇和半乳糖酸等半乳糖血症标志物的增高[5]。
3.2 成年发作II型瓜氨酸血症 Kobayashi等[10]报道本病发病年龄可以从11~79岁,其中以20~50岁为高发年龄段。男女比例接近2∶1,而Takenaka等[13]报道了42例CTLN2患者中男女发病率比为33∶9,平均发病年龄为34.7岁。总体上男性患病率高于女性。临床上主要表现为反复发作性的精神行为异常,伴不同程度的意识障碍甚至昏迷,起病急骤,可有身形消瘦表现。常由饮酒(该类患者不耐受酒精)、药物或感染所诱发。平素有偏食习惯,该类患者从小厌糖食却喜食富含蛋白质和脂类食物如花生、豆类、鸡蛋、鱼肉等。
高脂血症和肝癌是本病的主要并发症,约超过10%的患者合并有肝脏肿瘤。约有40%的患者开始被诊断为精神神经类疾病如癫痫、精神分裂症、抑郁症、睡眠病和帕金森氏病[14]。本病预后不良,常于发病几年后因脑水肿而死亡。
4 辅助检查
4.1 实验室检查 (1)生化检查:血有机酸代谢筛查可发现瓜氨酸水平升高,伴或不伴精氨酸水平升高。对于CTLN2,生化检查可发现血氨水平升高,通常为疾病发作期,并且以夜间显著升高为特点,而发作间期可能仅轻度升高或正常。(2)免疫组化检查:酶学检查可发现大多数患者肝脏特异性的精氨琥珀酸合成酶活性低下。但其肾脏及培养的成纤维细胞ASS活性、ASS基因及其mRNA无异常,据此可与经典型瓜氨酸血症,又名I型瓜氨酸血症鉴别。肝脏组织学检查见脂肪肝,但也发现肝纤维化及肝硬化。(3)基因测定:目前研究发现存在81种基因突变形式[15],包括错义突变、无义突变、插入及缺失突变等,其中以错义突变占据比例较大[16],而采用聚合酶链式反应技术进行基因序列的测定为诊断本病的金标准。
Komatsu等[17]通过对19例Citrin蛋白缺乏症的患者进行研究发现该类患者的体质量指数<20 kg/m2,血清胰分泌性胰蛋白酶抑制因子(pancreatic secretory trypsin inhibitor, PSTI)>29 ng/ml。其中89%的患者出现肝硬化表现,21%的患者曾被诊断为非酒精性脂肪性肝病(NAFLD),该病是Citrin缺陷相关的脂肪性肝炎。于是他们提出当临床医师遇到脂肪肝的患者同时却缺乏糖尿病、肥胖和代谢综合征表现,但既往曾有反复发作的胰腺炎病史或血清学检查提示PSTI水平升高者,要考虑Citrin缺陷病的可能。其中不饮酒而反复发作的慢性胰腺炎,是Citrin缺陷的一个重要指征。其中,PSTI主要由胰脏表达,但大多数CTLN2患者肝脏出现PSTI大量分泌和高效表达,提示血浆PSTI可作为CTLN2患者的特异性标志[18,19]。
4.2 影像学检查 头颅CT及MRI检查通常无特异性表现,可有脑萎缩性改变。急性起病的成人型患者早期,由于脑组织缺乏完整的尿素循环,血氨的升高则产生神经毒性,使脑星形细胞内谷氨酰胺合成增多,导致水分渗入细胞,从而引起脑水肿[20]。
5 诊断及鉴别诊断
Citrin蛋白缺乏症的诊断主要依据临床表现、实验室检查及基因测定等方面综合评估。其中以基因测定明确存在基因突变为诊断本病的金标准[21]。
本病主要与下列可引起血氨升高,引发类似于肝性脑病样症状的疾病鉴别:(1)瓜氨酸血症I型,即经典型瓜氨酸血症:本病由于ASS缺陷所致尿素循环受阻而引起高氨血症,属于常染色体隐性遗传性疾病,其致病基因位于第9号染色体长臂(9q34qter),主要的临床表现为呕吐、嗜睡、惊厥和昏迷等,通常在新生儿期起病,多在婴儿期死亡,多数患儿出生时无临床表现,24~72 h后出现上述临床症状,并且很快进展至呼吸衰竭及昏迷,病死率极高;(2)鸟氨酸氨甲酰转移酶缺乏症:是一种相对常见且十分严重的遗传代谢性疾病,由鸟氨酸甲酰基转移酶(ornithine carbamoyl transferase,OTC)基因突变引起,为X连锁隐性遗传病,临床表现为高氨血症及继发性的神经系统和肝脏损害,严重者在新生儿阶段出现呕吐、拒食、嗜睡、抽搐、昏迷以致死亡,通过血液串联质谱分析可进行诊断;(3)肝豆状核变性(hepatolenticular degeneration,HLD):由Wilson在1912年首先描述,故又称为Wilson病(WD),是一种常染色体隐性遗传性疾病,以铜代谢障碍引起的肝硬化、基底节损害为主的脑变性疾病为特点,以进行性加重的锥体外系症状、精神症状、肝硬化、肾功能损害及角膜色素环为主要表现,好发于青少年。上述疾病的实验室检查特点见表1。
6 治 疗
6.1 基本治疗 对于NICCD患儿,饮食方面可给予高蛋白、高脂、低碳水化合物饮食,注意补充必需氨基酸,包括精氨酸,有研究表明,不应限制或纠正其嗜食富含高脂、高蛋白而厌食谷类的饮食倾向。由于存在血半乳糖及短链、长链酰基肉碱增高,因此无乳糖或富含中链脂酰肉碱的配方奶被广泛应用于改善症状,苯巴比妥和熊脱氧胆酸用于黄疸对症处理,适当补充维生素E可改善氧化应激,有出血倾向患儿给予维生素K治疗。少数病情严重患儿需进行肝移植[22,23]。
对于CTLN2患者,因其存在明显的高氨血症,为减轻高血氨对神经系统的毒性作用,应长期坚持低碳水化合物饮食,可给予苯甲酸钠、苯乙酸钠等降低血氨。精氨酸可改善线粒体尿素循环酶的活性,因而可降低血氨水平,血氨显著升高时可进行血液或腹膜透析治疗[24,25]。
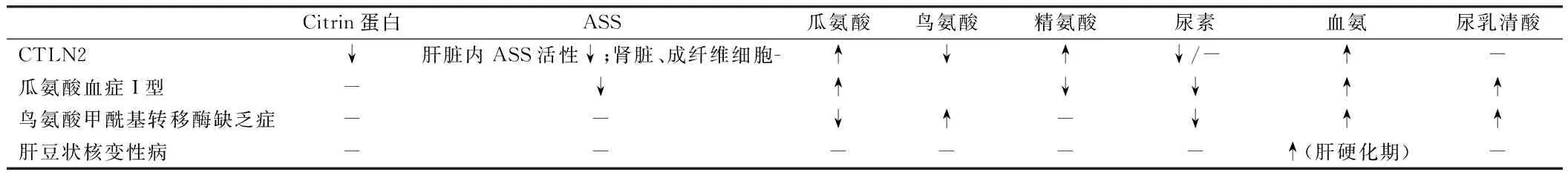
表1 血氨升高类疾病的实验室检查特点
注:ASS为精氨酸琥珀酸合成酶
有文献报道了3例CTLN2型患者在急性期时为减轻脑水肿而予以脱水降颅压治疗,其中2例因同时予以甘油及甘露醇治疗而引起脑水肿迅速进展,另1例则只输注了甘露醇而使症状得以缓解,推测输注甘油可引起细胞浆内NADH/NAD+比值的升高,影响了细胞的代谢从而使疾病加速进展,而甘露醇则对机体代谢的影响极小[6]。故对于伴有肝性脑病的CTLN2患者,使用甘油果糖会使体内NADH/NAD+比值升高,进一步加重高血氨和脑损伤,所以在减轻脑水肿时应选用甘露醇予以治疗。
6.2 肝移植 因本病病因为肝脏中特异性的Citrin蛋白缺乏所致,故上述保守治疗只能延缓病情的发展,并不能根治本病,目前对Citrin缺乏症惟一有效的治疗方法为肝移植,但对于移植后患者生存质量及时间缺乏相关研究,因此对肝移植后的效果尚难评价。
7 预 后
本病虽然与发现是否及时及长期治疗过程密切相关,但是临床分型不同,其预后相差很多。其中,NICCD病程多为自限性,预后较好。但也有患儿尽管经过积极治疗仍然发展为肝衰竭,最终需进行肝脏移植。在NICCD症状缓解后的代偿期,部分患者可以表现为健康状态,而受环境、饮食结构或个体素质等因素的影响,有些患者机体适应机制受损或代偿机制失调,部分患儿症状缓解十到数十年后发展为致命性的CTLN2,或者在症状缓解后出现低血糖酮症、智能发育的短暂落后,以及癫痫发作、胰腺炎、高蛋白饮食嗜好等其他类似于CTLN2的表现[26]。而成人型总体预后不良,从症状出现到死亡平均时间为26.4个月,71%的患者存活时间不超过2年[14]。
1 Kobayashi K,Sinasac DS,Iijima M,et al.The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein[J].Nat Genet,1999,22(2):159-163.
2 Kobayashi K,Bang LY,Xian LM,et al.Screening of nine SLC25A13 mutations: their frequency in patients with citrin deficiency and high carrier rates in Asian populations[J].Mol Genet Metab,2003,80(3):356-359.
3 Shigematsu Y,Hirano S,Hata I,et al.Newborn mass screening and selective screening using electrospray tandem mass spectrometry in Japan[J].J Chromatogr B Analyt Technol Biomed Life Sci,2002,776(1):39-48.
4 Saheki T,Inoue K,Tushima A,et al.Citrin deficiency and current treatment concepts[J].Mol Genet Metab,2010,100(Suppl 1):S59-S64.
5 Saheki T,Kobayashi K.Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD)[J].J Hum Genet,2002,47(7):333-341.
6 Yazaki M,Takei Y,Kobayashi K,et al.Risk of worsened encephalopathy after intravenous glycerol therapy in patients with adult-onset type II citrullinemia (CTLN2)[J].Intern Med,2005,44(3):188-195.
7 Saheki T,Kobayashi K,Iijima M,et al.Adult-onset type II citrullinemia and idiopathic neonatal hepatitis caused by citrin deficiency: involvement of the aspartate glutamate carrier for urea synthesis and maintenance of the urea cycle[J].Mol Genet Metab,2004,81(Suppl 1):S20-S26.
8 Yeh JN,Jeng YM,Chen HL,et al.Hepatic steatosis and neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) in Taiwanese infants[J].J Pediatr,2006,148(5):642-646.
9 Ohura T,Kobayashi K,Tazawa Y,et al.Neonatal presentation of adult-onset type II citrullinemia[J].Hum Genet,2001,108(2):87-90.
10 Abukawa D,Ohura T,Iinuma K,et al.An undescribed subset of neonatal intrahepatic cholestasis associated with multiple hyperaminoacidemia[J].Hepatol Res,2001,21(1):8-13.
11 Tomomasa T,Kobayashi K,Kaneko H,et al.Possible clinical and histologic manifestations of adult-onset type II citrullinemia in early infancy[J].J Pediatr,2001,138(5):741-743.
12 Ohura T,Abukawa D,Aikawa J,et al.Disturbance of amino acids metabolism in infants with idiopathic neonatal hepatitis[J].Jpn J Pediatr Soc,1997,101:1522-1525.
13 Takenaka K,Yasuda I,Araki H,et al.Type II citrullinemia in an elder patient treated with living related partial liver transplantation[J].Intern Medicine,2000,39(7):553-558.
14 Saheki T, Kobayashi K. Mitochondrial aspartate glutamate carrier(citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD)[J].J Hum Genet,2002,47(7):333-341.
15 Song YZ,Zhang ZH,Lin WX,et al.SLC25A13 gene analysis in citrin deficiency: sixteen novel mutations in East Asian patients, and the mutation distribution in a large pediatric cohort in China[J].PLoS One,2013,8(9):e74544.
16 Zhang ZH,Lin WX,Deng M,et al.Clinical, molecular and functional investigation on an infant with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD)[J].PLoS One,2014,9(2):e89267.
17 Komatsu M,Yazaki M,Tanaka N,et al.Citrin deficiency as a cause of chronic liver disorder mimicking non-alcoholic fatty liver disease[J].J Hepatol,2008,49(5):810-820.
18 Tsuboi Y,Fujino Y,Kobayashi K,et al.High serum pancreatic secretory trypsin inhibitor before onset of type II citrullinemia[J].Neurology,2001,57(5):933.
19 Kobayashi K,Horiuchi M,Saheki T.Pancreatic secretory trypsin inhibitor as a diagnostic marker for adult-onset type II citrullinemia[J].Hepatology,1997,25(5):1160-1165.
20 Rama Rao KV,Norenberg MD.Glutamine in the pathogenesis of hepatic encephalopathy: the Trojan horse hypothesis revisited[J].Neurochem Res,2014,39(3):593-598.
21 Häberle J,Boddaert N,Burlina A,et al.Suggested guidelines for the diagnosis and management of urea cycle disorders[J].Orphanet J Rare Dis,2012,7:32.
22 Nagasaka H,Okano Y,Tsukahara H,et al.Sustaining hypercitrullinemia, hypercholesterolemia and augmented oxidative stress in Japanese children with aspartate/glutamate carrier isoform 2-citrin-deficiency even during the silent period[J].Mol Genet Metab,2009,97(1):21-26.
23 Saheki T,Kobayashi K,Terashi M,et al.Reduced carbohydrate intake in citrin-deficient subjects[J].J Inherit Metab Dis,2008,31(3):386-394.
24 Mutoh K,Kurokawa K,Kobayashi K,et al.Treatment of a citrin-deficient patient at the early stage of adult-onset type II citrullinaemia with arginine and Sodium pyruvate[J].J Inherit Metab Dis,2008,31(Suppl 2):S343-S347.
25 Hayasaka K,Numakura C,Toyota K,et al.Treatment with lactose (galactose)-restricted and medium-chain triglyceride-supplemented formula for neonatal intrahepatic cholestasis caused by citrin deficiency[J].JIMD Rep,2012,2:37-44.
26 Ohura T,Kobayashi K,Tazawa Y,et al.Clinical pictures of 75 patients with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD)[J].J Inherit Metab Dis,2007,30(2):139-144.
100053 北京,首都医科大学宣武医院神经内科
王向波,E-mail:xb90956@sina.cn
10.3969 / j.issn.1671-6450.2014.09.039
2014-05-04)
猜你喜欢
今日农业(2021年16期)2021-11-26
现代畜牧科技(2021年6期)2021-07-16
现代畜牧科技(2021年6期)2021-07-16
猪业科学(2018年5期)2018-07-17
环球时报(2018-04-19)2018-04-19
中外文摘(2017年15期)2017-11-13
现代检验医学杂志(2016年1期)2016-11-12
中国免疫学杂志(2016年2期)2016-01-30
疑难病杂志(2014年12期)2014-04-16
大众文艺(2014年5期)2014-03-12
