
脑室周围白质软化症再髓鞘化的研究进展
2014-08-09 01:29陈鹏慧
遵义医科大学学报 2014年1期
陈鹏慧
(第三军医大学 神经生物学教研室,重庆 400038)
脑室周围白质软化(periventricular leukomalacia,PVL)是一种发生于围生期早产儿的进行性疾病,主要损伤在大脑皮层的白质区域。PVL多发生在胎龄小于32周、出生体质量小于1500g的早产儿。常有宫内缺氧缺血、感染、绒毛羊膜炎、胎膜早破、窒息、低碳酸血症、反复呼吸暂停、心动过缓以及败血症等病史[1-2]。患儿的临床症状早期可表现为下肢肌张力减低、颈部伸肌张力升高、呼吸暂停、心律过缓、激惹以及因假性延髓麻痹所致喂养困难等非特异性症状[2]。晚期可导致脑性瘫痪、视听和认知障碍等后遗症。其中,PVL患儿所表现的视力障碍并非早产儿视网膜病变,其视力受损主要与视神经髓鞘化障碍有关[3]。根据细胞生物学特点和损伤的病理表现可将PVL分为以局部少突胶质细胞及其前体细胞急性坏死为主的局灶性病变,和以少突胶质细胞谱系凋亡为主的弥散性病变。局灶性PVL位于脑白质深部,形成囊性病变,特征表现为所有细胞成分的局灶性凝固性坏死;而弥散性PVL主要表现为少突胶质细胞弥散性损伤,以及后续发生的髓鞘发育延迟、白质容积减少及脑室扩大[4]。
1 少突胶质细胞系的易损性是导致PVL主要原因
目前认为PVL的细胞学基础是神经元轴突发生脱髓鞘。主要原因在于白质区域少突胶质细胞系的易损性,尤其是未成熟的成髓鞘前少突胶质细胞(premyelinating oligodendrocyte,Pre-OLs)损伤,使PVL后期髓鞘再生难以进行[5]。Pre-OLs对缺氧、缺血、炎症等各种因素的高度敏感性和易损性,是引起PVL的主要病理生理学因素,均可导致髓鞘化障碍[6]。Pre-OLs的敏感性和易损性主要是由于其成熟程度所决定的。动物实验表明围生期缺氧缺血(Hyperxia ischemia,HI)可导致晚期少突胶质细胞前体细胞大量凋亡,而发育成熟脑内少突胶质细胞则对HI呈明显抵抗性,提示围生期少突胶质细胞谱系的易损性是依赖于脑发育的成熟度。晚期Pre-OLs是PVL病变的主要靶细胞,其机制可能与线粒体损伤、抗氧化防御缺陷及其反应的延迟、谷氨酸受体过度表达及谷氨酸逆转运有关等有关[7-8]。同时,氧自由基介导的白质损伤,兴奋性氨基酸的毒性作用,感染、炎症反应以及细胞因子的诱导等,也是引起Pre-OLs的易损性和进行性脑损伤从而导致PVL的重要原因[9]。
2 髓鞘再生的调控因素
中枢神经系统发生脱髓鞘后会通过不同的机制自我修复,主要途径是诱导少突胶质前体细胞(oligodendrocyte precursor cells,OPCs)向少突胶质细胞分化从而形成再髓鞘化[10]。这一过程受不同转录因子的调控、miRNA的作用以及表观遗传学组蛋白去乙酰化酶的调节来实现。其中,髓磷脂基因调节因子(myelin gene regulatory factor,MRF)是使Pre-OLs向成熟少突胶质细胞过渡的最主要的调节转录因子[11]。但是在PVL发生后,这种髓鞘化难以完全实现,使得PVL后髓鞘不能够得到有效的修复。反复脱髓鞘改变,影响轴突传递功能,进而可导致神经元坏死[12]。目前研究表明再髓鞘化过程中鞘磷脂基因的表达时空顺序非常关键,均在PVL中发挥不同作用[13]。
2.1 转录前的调控 在中枢神经系统的发育过程中,少突胶质细胞谱系起源于神经管腹侧的室管膜下区,在细胞分化中有许多因子参与并发挥重要作用(见图1)[14]。
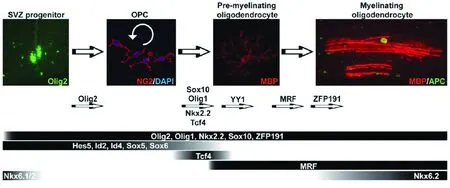
图1 Pre-OLs细胞分化为少突胶质细胞并形成髓鞘的主要调控因子[14]
在室下区的神经祖细胞向少突胶质谱系前体的OPCs的分化过程中,表达Nkx6.1、Nkx6.2 和Olig2等多种转录因子,其中Olig2对OPCs的分化具有决定性作用[15]。血小板衍生生长因子α受体(platelet derived growth factor α receptor,PDGFRα)是OPCs的特征性标志物之一,研究发现虽然在Olig2-/-基因敲除小鼠脑内不表达PDGFRα免疫反应阳性细胞,但是在Olig2+的小鼠中用基因干扰下调Nkx6.1和Nkx6.2的表达,OPCs向少突胶质细胞的分化并未受到显著干扰[16]。一旦少突胶质细胞分化确定,并实现其主要生物学作用,其谱系的其他转录因子则主要发挥维持OPCs增殖特性而抑制其再分化的作用,这些因子包括Sox5、Sox6,以及bHLH蛋白家族的Hes5、Id2和Id4等[17]。在OPCs分化为Pre-OLs的过程中,Nkx2.2、Sox10和 Olig1在早期即开始表达,但是这些因子对于细胞的特化以及迁移都没有作用,而是仅仅参与向成熟少突胶质细胞的分化和调节鞘磷脂基因的表达,以建立髓鞘。在Sox10-/-小鼠中虽然可以用过表达Sox9功能的方法,使基因缺陷小鼠OPCs数量正常,但是这些细胞在分化后期却不能表达髓鞘标志分子——髓鞘碱性蛋白(myelin basic protein,MBP),也即不能进一步分化成髓鞘的功能性细胞[17]。同样,在Nkx2.2敲除的小鼠中亦未发现明显的OPCs数量减少,而显示MBP表达阴性。反之,在Olig2+小鼠中成功诱导Nkx2.2表达后,又发现大量MBP表达[18]。表明Nkx2.2是少突胶质细胞系中形成功能性髓鞘的必须因子。
此外,虽然Tcf4在整个少突胶质细胞分化成熟的过程中只是一过性的表达,但是Tcf4与Wnt/β-catenin信号通路的TCF4/β-catenin或Groucho/Tle家族成员形成不同的复合体发挥不同作用[19]。TCF4/β-catenin形成以后,主要通过下游的靶分子Id2和Id4发挥作用,使OPCs脱离原来的细胞周期进入分化过程[20]。其他研究则发现通过不同的联合机制使TCF4不表达,如上调β-catenin的拮抗剂APC、组蛋白去乙酰化及Groucho/Tle 1竞争结合TCF4等,在TCF4/β-catenin解离后发现也可以促进OPCs的分化以及鞘磷脂基因的表达[21]。因此,Tcf4在OPCs的分化及鞘磷脂形成中究竟是起正向还是负向调节作用尚有待明确。
在少突胶质细胞分化成熟的后期,ZFP191发挥重要的作用,其表达异常或功能异常可导致严重的髓鞘化障碍。在ZFP191缺失的少突胶质细胞中,细胞停留在相对较晚期的未成熟前体细胞阶段,不能够继续发育为成熟的具有成髓鞘功能的少突胶质细胞,这是与Olig1、Sox10 和MRF等其他的转录因子显著的区别[22]。
2.2 转录后的调控 新近研究发现,除转录因子外,少突胶质细胞谱系的分化成熟也受许多miRNA调控。miRNA是一种小的非编码RNA,首次发现于秀丽隐杆线虫,随后发现miRNA是一种比较保守的转录后调节机制,在包括哺乳动物在内的不同种系都发挥重要作用[23]。在转录过程中,DNA转录形成初级转录本Pri-miRNA,在细胞核内经Drosha酶处理后形成具有延伸发夹结构的原始miRNA,或称为Pre-miRNA。后者转运到细胞核外,经Dicer酶处理后形成具19-25个核苷酸的双链RNA,成为miRNA诱导沉默复合体(miRNA-induced silencing complex,RISC)的一部分,RISC通过与mRNA互补的单链结构来调节许多mRNA的表达[24]。RISC在细胞内主要作用是抑制mRNA表达,所以miRNA具有与许多其他转录因子不同的作用,miRNA的多样性能够控制复杂的细胞活动。
基因芯片分析少突胶质细胞miRNA,发现miR-219和miR-338在少突胶质细胞的分化成熟过程中发挥重要作用。对培养的少突胶质前体细胞敲除Drosha酶,使其转录过程不能产生pre-miRNA,发现培养细胞亦不能够正常地分化成熟的有髓鞘化功能的少突胶质细胞。而再转染miR-219 或miR-338后,可以部分解除分化和成熟抑制作用(见图2)[14]。在发育期动物脑内立体定位注射过量的miR-219和miR-338,发现有MBP的提前表达[25]。这说明miR-219和miR-338是成熟少突胶质细胞分化的重要因子。此外,miR-219 和miR-338还具有调节脂质代谢基因的功能,这与少突胶质细胞分化成熟后期鞘磷脂基因的表达密切相关。通过对这两种miRNA目标序列进行生物信息学筛查研究发现,miR-219主要与许多目标基因mRNA的3’UTR结合发挥作用,这些分子主要包括:Hes5、Sox6、Zfp238 和FoxJ3等[26]。
2.3 表观遗传学的调控 除了复杂的转录调节因子调节少突胶质细胞系的分化成熟外,还存在表观遗传学机制,主要表现为组蛋白去磷酸化酶家族的多重调节作用。一般情况下,组蛋白的乙酰化有利于DNA与组蛋白八聚体的解离,核小体结构松弛,从而使各种转录因子和协同转录因子与DNA结合位点特异性结合,激活基因的转录。而HDAC可通过染色体的结构修饰和调控相关基固的表达,对髓磷脂基因转录后表达有重要调节作用[27]。
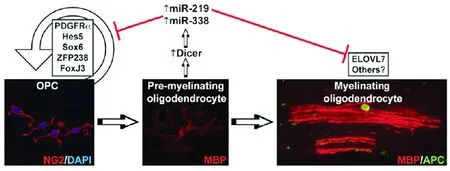
图2 不同miRNA对少突胶质细胞分化的调节作用[14]
应用HDAC拮抗剂曲古菌素A和丙戊酸钠对组蛋白去磷酸化酶家族筛查,发现HDAC通过多种机制调节少突胶质细胞系的分化成熟过程,HDAC也是OPCs分化必须因素。在体研究发现大鼠胼胝体髓鞘化需要HDAC的作用[28]。对出生后小鼠大脑的发育后期研究,观察到组蛋白标记物H3K9me3和异染色质蛋白的表达量大量减少。因此推测HDAC主要通过抑制多种髓鞘化抑制因子的转录而发挥作用。但是一旦抑制的时空性结束,髓鞘化过程启动后,HDAC的作用就不再是该过程的必要条件[29]。
HDAC可促进OPCs向少突胶质细胞系的顺行分化。反之,抑制HDAC可使OPCs逆分化为神经干细胞样细胞,这种细胞再分化时可以形成神经元。因此,HDAC可能是少突胶质细胞跨系分化的因素之一。进一步研究发现这些细胞内干细胞分子标记物如Sox2等在逆分化时表达上调,这可能是抑制HDAC的作用后,染色体的松解使得Sox2启动子被激活而重新大量表达。与此同时还可观察到许多其他干细胞基因的表达也上调,而成熟少突胶质细胞系的基因表达下调[30]。如果在神经干细胞中过量表达HDAC1,HDAC2或HDAC3,在胚胎分化后期OPCs向少突胶质细胞系的分化能力显著加强[31]。少突胶质细胞系的分化也受Wnt/ABC信号通路的抑制。通过杂交表达Cre重组酶条件控制Olig1的小鼠和带有floxed Hdac1和 floxed Hdac2基因的小鼠,使少突胶质细胞中Hdac1和Hdac2同时沉默后发现,在双重突变小鼠中,OPCs的生成和分化都严重受损,而ABC的表达量却大幅度上调,当HDAC1和HDAC2都不表达时,可促进OPCs分化[31]。HDAC1和 HDAC2还可以与SMRT相互作用而抑制Notch信号通路促进OPCs分化。HDAC家族的其他成员如HDAC6和HDAC11等也可以通过类似的方式调节OPCs的成熟分化[32]。
3 MRF是调节髓鞘再生的关键因子
髓磷脂基因调节因子MRF是少突胶质细胞特异性的DNA结合蛋白,其编码基因位于1号染色体长臂4带3区上。运用流式细胞技术结合基因分析的方法对中枢神经系统内特异性的基因进行筛查研究,发现MRF是少突胶质细胞谱系所特有的基因[10-11]。进而用MRF寡核苷酸探针对脑内不同的细胞进行标记,神经元和星形胶质细胞均未检测到标记探针的阳性反应,在OPCs探针标记表达也非常少,而在分化后的少突胶质细胞中随着细胞的成熟度,MRF表达量逐渐增加[33]。
培养的少突胶质细胞如果敲除MRF会阻止鞘磷脂相关基因的表达。反之,体内或者体外使MRF基因强制表达后会诱导鞘磷脂基因的表达。在培养的OPCs中转染针对MRF基因靶向性的siRNA后发现,OPCs可以表达PDGFRα和NG2等标记物,也可以正常分化为Pre-OLs,但是却不能继续分化成熟且表达髓鞘标志分子MBP,这说明MRF主要在发育后期Pre-OLs的分化和形成成髓鞘少突胶质细胞中发挥作用[34]。
在条件敲除MRF的小鼠,少突胶质细胞谱系细胞可以正常分化,但是却不能够表达鞘磷脂基因,以后因髓鞘化障碍细胞趋于凋亡。MRF只在分裂后的Pre-OLs中表达,是鞘磷脂基因表达所特需的,而不是少突胶质细胞谱系特化所需,这与其他因子如Nxk2.2、Olig1/2、Sox10和YY1等在OPCs和少突胶质细胞均表达有所不同[17,35]。中枢神经系统MRF是与周围神经系统的Krox-20具有同样重要性的调节鞘磷脂基因表达的转录调节因子。Krox-20在周围神经系统中参与调节未成熟的施万细胞向成熟施万细胞过渡,因此在周围神经髓鞘化的过程中发挥至关重要的作用[36]。而Krox-20由YY1诱导产生,研究发现中枢神经系统MRF基因的内含子也具有与YY1相同的位点,这说明YY1可能也参与中枢神经系统诱导MRF的表达[37]。
总之,作为髓磷脂的重要调节因子,MRF作用途径和机制主要有:①直接作为少突胶质细胞一个转录因子调节鞘磷脂基因的表达;②与其他的调节因子一起协同诱导鞘磷脂基因的表达,或者发挥调控作用影响其他鞘磷脂基因转录子的表达;③直接结合在鞘磷脂基因的启动子上调节其表达,使少突胶质细胞细胞过渡到成髓鞘的成熟性状态;④参与调节少突胶质细胞谱系细胞其他转录因子功能。
4 展望
PVL是各种原因导致的主要以损伤大脑白质为主的新生儿疾病,Pre-OLs因在围生期对缺血缺氧的高度敏感性和易损性,成为新生儿脑损伤的靶细胞。发育期OPCs分化为Pre-OLs并成为成熟的成髓鞘少突胶质细胞,对损伤后启动自身修复有重要的作用,其成熟、分化和再髓鞘化过程也受转录因子、几种miRNA和HDAC的调节。其中MRF作为重要的转录因子,在Pre-OLs分化成为功能性的少突胶质细胞这一阶段发挥非常重要的作用,对其机制的深入研究,可望成为脑室周围白质软化症及脑瘫预后的新靶点。
[参考文献]
[1] Resch B, Resch E,Maurer U,et al.Periventricular leukomalacia and neurodevelopmental outcome[J].J Pediatr,2011,159(6):1049-1050.
[2] Imamura T, Ariga H, Kaneko M, et al. Neurodevelopmental outcomes of children with periventricular leukomalacia[J].Pediatr Neonatol,2013,54(6):367-372.
[3] Lehman S S. Cortical visual impairment in children: identification, evaluation and diagnosis[J].Curr Opin Ophthalmol,2012,23(5):384-387.
[4] de Bruïne F T,van den Berg-Huysmans A A, Leijser L M,et al.Clinical implications of MR imaging findings in the white matter in very preterm infants: a 2-year follow-up study[J]. Radiology, 2011, 261(3):899-906.
[5] Volpe J J, Kinney H C,Jensen F E,et al.The developing oligodendrocyte: key cellular target in brain injury in the premature infant[J].Int J Dev Neurosci,2011,29(4):423-440.
[6] Falahati S, Breu M, Waickman A T,et al. Ischemia-induced neuroinflammation is associated with disrupted development of oligodendrocyte progenitors in a model of periventricular leukomalacia[J].Dev Neurosci,2013,35(2-3):182-196.
[7] Fields R D.Glutamate receptors: the cause or cure in perinatal white matter injury[J].Neuron Glia Biol, 2010,6(4):209-211.
[8] Jantzie L L, Talos D M, Jackson M C,et al.Developmental Expression of N-Methyl-D-Aspartate (NMDA) Receptor Subunits in Human White and Gray Matter: Potential Mechanism of Increased Vulnerability in the Immature Brain[J].Cereb Cortex,2013,bht246vl-bht246.
[10] Emery B, Agalliu D, Cahoy J D,et al. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination[J]. Cell, 2009, 138(1):172-185.
[11] Koenning M, Jackson S, Hay C M, et al. Myelin gene regulatory factor is required for maintenance of myelin and mature oligodendrocyte identity in the adult CNS[J].J Neurosci,2012,32(36):12528-12542.
[12] Kinney H C, Haynes R L, Xu G, et al. Neuron deficit in the white matter and subplate in periventricular leukomalacia[J].Ann Neurol,2012,71(3):397-406.
[13] Emery B. Regulation of oligodendrocyte differentiation and myelination[J].Science,2010,330:779-782.
[14] Emery B. Transcriptional and post-transcriptional control of CNS myelination[J].Curr Opin Neurobiol,2010,20(5):601-607.
[15] Panman L, Andersson E, Alekseenko Z,et al. Transcription factor-induced lineage selection of stem-cell-derived neural progenitor cells[J].Cell Stem Cell,2011,8(6):663-675.
[16] Liu Y, Jiang P, Deng W. OLIG gene targeting in human pluripotent stem cells for motor neuron and oligodendrocyte differentiation[J].Nat Protoc,2011, 6(5):640-655.
[17] Küspert M, Hammer A, Bösl M R, et al. Olig2 regulates Sox10 expression in oligodendrocyte precursors through an evolutionary conserved distal enhancer[J].Nucleic Acids Res,2011,39(4):1280-1293.
[18] Gotoh H, Ono K, Takebayashi H, et al. Genetically-defined lineage tracing of Nkx2.2-expressing cells in chick spinal cord[J].Dev Biol,2011,349(2):504-511.
[19] Haines J D, Fang J, Mushynski W E, et al. Mitogen-activated protein kinase activated protein kinase 2 (MK2) participates in p38 MAPK regulated control of oligodendrocyte differentiation[J].Glia,2010,58(11):1384-1393.
[20] Chew L J, Shen W, Ming X, et al. SRY-box containing gene 17 regulates the Wnt/β-catenin signaling pathway in oligodendrocyte progenitor cells[J].J Neurosci,2011,31(39):13921-13935.
[21] Tawk M, Makoukji J, Belle M, et al. Wnt/beta-catenin signaling is an essential and direct driver of myelin gene expression and myelinogenesis[J].J Neurosci,2011,31(10):3729-3742.
[22] Howng S Y,Avila R L,Emery B, et al. ZFP191 is required by oligodendrocytes for CNS myelination[J].Genes Dev,2010,24(3):301-311.
[23] Barca-Mayo O, Lu Q R. Fine-Tuning Oligodendrocyte Development by microRNAs[J].Front Neurosci,2012,6:13.
[24] Zhao X, Wu J, Zheng M,et al. Specification and maintenance of oligodendrocyte precursor cells from neural progenitor cells: Involvement of microRNA-7a[J].Mol Biol Cell,2012,23(15):2867-2878.
[25] de Faria O Jr, Cui Q L, Bin J M, et al.Regulation of miRNA 219 and miRNA clusters 338 and 17-92 in oligodendrocytes[J]. Front Genet,2012,3:46.
[26] Dugas J C,Cuellar T L, Scholze A, et al. Dicer1 and miR-219 are required for normal oligodendrocyte differentiation and myelination[J]. Neuron, 2010, 65(5):597-611.
[27] Koch M W, Metz L M, Kovalchuk O. Epigenetic changes in patients with multiple sclerosis[J].Nat Rev Neurol,2013, 9(1):35-43.
[28] Conway G D,O'Bara M A, Vedia B H,et al. Histone deacetylase activity is required for human oligodendrocyte progenitor differentiation[J].Glia,2012,60(12):1944-1953.
[29] Shen S, Sandoval J, Swiss V A, et al.Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency[J].Nat Neurosci,2008,11(9):1024-1034.
[30] Revet I, Feeney L, Tang A A, et al.Dysmyelination not demyelination causes neurological symptoms in preweaned mice in a murine model of Cockayne syndrome[J].Proc Natl Acad Sci USA,2012,109(12):4627-4632.
[31] Ye F, Chen Y, Hoang T, et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction[J].Nat Neurosci,2009,12(7):829-838.
[32] Swiss V A,Nguyen T, Dugas J,et al. Identification of a gene regulatory network necessary for the initiation of oligodendrocyte differentiation[J].PLoS One,2011,6(4):e18088.
[33] Plemel J R, Manesh S B,Sparling J S,et al. Myelin inhibits oligodendroglial maturation and regulates oligodendrocytic transcription factor expression[J].Glia,2013,61(9):1471-1487.
[34] Mela A, Goldman J E.CD82 blocks cMet activation and overcomes hepatocyte growth factor effects on oligodendrocyte precursor differentiation[J].J Neurosci,2013,33(18):7952-7960.
[35] 井秀杰,曹云涛. 少突胶质细胞相关神经抑制因子[J].遵义医学院学报,2012,35(4):347-349.
[36] Fröb F, Bremer M, Finzsch M, et al. Establishment of myelinating schwann cells and barrier integrity between central and peripheral nervous systems depend on Sox10[J].Glia,2012,60(5):806-819.
[37] Hossain S, de la Cruz-Morcillo M A, Sanchez-Prieto R, et al. Mitogen-activated protein kinase p38 regulates Krox-20 to direct Schwann cell differentiation and peripheral myelination[J].Glia,2012,60(7): 1130-1144.
猜你喜欢
中华耳科学杂志(2022年1期)2022-11-24
磁共振成像(2022年8期)2022-10-08
神经损伤与功能重建(2020年10期)2020-12-23
神经损伤与功能重建(2020年11期)2020-12-01
中国医学影像技术(2020年8期)2020-01-13
党的生活(黑龙江)(2018年9期)2018-10-17
飞碟探索(2015年11期)2015-09-10
中国生化药物杂志(2015年4期)2015-07-07
医学研究杂志(2015年9期)2015-07-01
癌变·畸变·突变(2015年4期)2015-02-27
