
氧化石墨烯自组装水凝胶
2014-08-07 08:22黄志锋
厦门大学学报(自然科学版) 2014年5期
黄志锋,李 磊,白 华
(厦门大学材料学院,福建 厦门 361005)
在基于GO材料中,自组装水凝胶是一类独特的材料.水凝胶是一种特殊的分散体系,是由高分子、小分子聚集体或胶体粒子通过物理或者化学结合力形成的三维网络结构,在网络内部的空隙中则充满水.水凝胶具有良好的生物相容性、负载性能、渗透性.而在超分子水凝胶中,三维网络的结合力为可逆的超分子作用,这赋予了水凝胶可逆性与自修复性等新的性能.目前,水凝胶在药物负载与释放、高吸水树脂、污染物吸附、传感等领域有着日益重要的应用.有关GO凝胶化的报道最早见于2009年,Luo[12]发现超大尺寸GO在很低的质量分数下(0.3%)失去流动性,形成水凝胶(如图1(A)所示),但作者并未进一步对水凝胶加以表征研究.此后,Bai等[13]首次报道了普通GO在微量聚乙烯醇(PVA)的作用下,形成超分子复合水凝胶.这一工作开辟了GO自组装复合水凝胶的研究领域.在传统凝胶体系中,三维网络通常是由一维的分子或分子聚集体(纳米纤维)所组成,而在这些GO水凝胶中,网络是由二维结构单元聚集而成(见图1(B)).因此,GO水凝胶的形成与结构都与传统水凝胶有较大的区别,对GO水凝胶的研究可以丰富凝胶化学的理论.此外,GO具有多样的表面基团和巨大的比表面积,这些性质在水凝胶中得以保留,极大地拓展了GO的应用.最近一段时期,GO自组装水凝胶得到了快速发展.
本文的目的即为总结近期GO超分子水凝胶的研究进展.所讨论的对象限于以GO为主要凝胶因子,通过超分子作用交联形成的水凝胶.文献中还存在另一种GO复合水凝胶,其中GO作为纳米填料添加在其他聚合物中形成的凝胶.由于这类水凝胶与GO自组装水凝胶的结构和性质有本质的区别,所以本文不涉及此类凝胶.在本文中,我们在分析GO化学结构的基础上,总结GO水凝胶的制备方法和形成机理,着重讨论形成凝胶的超分子作用力,包括氢键、静电作用、配位作用、π-π作用等.之后,我们总结了GO自组装水凝胶的应用,并展望该领域今后的发展方向.

图1 超大尺寸GO在0.3%的质量分数下所形成的 水凝胶[12](A);GO与聚乙烯基吡咯烷酮(PVP)形成的 复合水凝胶中的三维网络结构(标尺:10 μm)[13](B)Fig.1 A hydrogel with 0.3% large GO sheets as gelator[12] (A);Three-dimensional network in the GO/poly(vinyl pyrrolidone) composite hydrogel (scale bar:10 μm)[13] (B)
1 GO的制备及结构
GO是通过强氧化剂氧化石墨类原料而制备的.使用最广泛的原料为天然石墨,除天然石墨之外[14],可以用于制备GO的原材料还包括膨胀石墨、石墨烯等[12,15].石墨的氧化反应通常在强酸性条件下进行,最为常用的方法是Hummers法[14].该方法在浓硫酸中,使用高锰酸钾作为氧化剂,在硝酸钠的存在下,氧化天然石墨,得到氧化石墨.氧化石墨在超声或搅拌的作用下剥离,以单层的状态分散在水中,形成GO分散液.在反应过程中,石墨片层的共轭结构被氧化,生成含氧基团;同时石墨片层中部分碳碳键断裂[16],导致GO的纳米片上存在一些空洞,片的尺寸也明显小于原料石墨的颗粒尺寸.除高锰酸钾之外,可以用于氧化天然石墨的氧化剂还包括氯酸钾等[17].
虽然上述合成GO的反应早在一百多年前已经被发现,但GO的结构还没有完全确定.根据GO的元素组成(~ C2O,及少量氢),人们曾提出过多种GO的化学结构,目前广泛接受的结构是基于Lerf-Klinowski所建立的模型,如图2所示[18].GO上存在多种基团,包括:小面积的共轭区域、羟基、环氧基、羰基、羧基、内酯基团等[19].Cai等[15]根据二维核磁数据推断,环氧基团和羟基处于相邻的位置;Gao等[19]也通过对核磁碳谱的仔细研究,发现GO上存在五元和六元内酯基团的证据.值得注意的是,GO的具体结构与其制备条件有密切的关系.不同实验室在不同条件下制备的GO的氧化程度可能存在差异,因此其化学组成不完全相同.此外,新制的GO的结构不稳定,在储存过程中,还可能发生一些复杂的反应.Dimiev等[20]发现,新制的GO主要含有环氧基团和少量羟基,以及部分硫酸酯.这种GO与水接触会发生复杂水解反应,形成邻二醇结构,同时部分脱氧恢复共轭结构.GO的酸性即来源于未水解的、共价连接在GO上的单硫酸酯.尽管学者对GO的结构细节存在不同的观点,但GO中环氧基团和邻二羟基结构的存在已被不同实验室中获得的波谱数据所证实,得到公认.
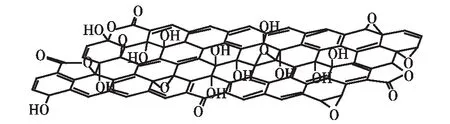
图2 GO的可能结构[18]Fig.2 Possible structure of GO[18]
从化学结构来看,GO上存在的这些官能团能赋予GO多种超分子相互作用.例如:GO中的羟基、环氧基、羧基、羰基、酯基等都能形成氢键,其中羟基既是氢键给体又是氢键受体;电离的羧基和硫酸基团带有负电荷,可以提供静电作用;岛状的共轭区域具有大π键,能提供π-π相互作用和疏水作用;而羟基、羧基、羰基等基团可以与金属离子配位.这些超分子相互作用在GO自组装的过程中起到了关键作用,正是由于这些超分子作用的存在,GO才能与多种物质形成自组装水凝胶.
2 GO自组装水凝胶的制备方法
GO自组装水凝胶的制备比较简单.通常,一定浓度的GO溶液与另一种能与GO作用的物质(超分子交联剂)的溶液混合震荡[13],则可以自发地形成水凝胶.水凝胶的形成可以通过试管倒置法监测,动态流变数据也能体现出,水凝胶的储能模量远远高于初始GO溶液[13].在某些情况下,由于GO和交联剂的作用很强,在二者溶液接触的瞬间形成聚集体,无法得到均匀的水凝胶.此时可以利用超分子凝胶的可逆性,通过扩散的方法制备水凝胶.例如,壳聚糖(CS)溶液与GO溶液在碱性条件下混合不能形成水凝胶,但是上述混合溶液在盐酸溶液中透析,使盐酸扩散进入混合溶液,则由于pH值下降,溶液转化为均匀的水凝胶[21].此外,原位聚合也可以用于制备GO与聚合物的复合水凝胶.例如,在GO溶液中进行导电聚合物(CP)的氧化聚合,可以得到具有导电性复合水凝胶[22].
专项扣除指的就是按照纳税人不同的情况和社会的发展情况,进而设计的特殊扣除,其中包括住房支出,在教育方面的支出,赡养方面的支出,医疗保健方面的支出这几个方面的专项扣除。如今在世界上大多数的国家都开始落实分类和综合相联系的个人所得税制,而且都存在专项扣除项目。在我们国家今年的政府报告中明确提出了增加个税专项扣除之后,这次个税法修正案草案里面明确规定,在划分综合所得征收的时候,需要增加子女教育支出部分,租房租金部分以及住房贷款等部分的附加扣除。
3 GO自组装水凝胶的形成机理
GO水凝胶可以在很低的浓度下形成,通常临界凝胶浓度在0.3%(质量分数,下同) 左右.其强度与交联剂(或交联作用)的类型有关,对GO质量分数为0.5%的水凝胶来说,储能模量可以从102Pa到105Pa不等.这种较低的临界凝胶浓度和较高的力学强度,表明GO作为一种二维的凝胶因子,其凝胶行为与传统聚合物水凝胶不同.
凝胶本质上是由凝胶因子所构成的三维网络,所以GO溶液的凝胶化过程,即为GO片自组装、聚集形成三维网络的过程.然而GO在水中具有良好的溶解性,通常不易发生聚集.这是因为:1) GO表面含有很多亲水基团,水合能较大;2) GO上酸根电离产生负电荷,静电排斥作用阻止GO片的聚集.因此,为了制备GO水凝胶,必须增强GO片的结合力,或者削弱GO之间的排斥力,使其在水溶液中的稳定性下降,发生聚集.超分子交联剂的作用即通过提供超分子作用力来增强GO片之间的结合力.需要强调的是,水溶液中的物质发生聚集不一定会形成水凝胶,还可能以沉淀的形式在水中析出.因此,GO独特的二维形状,必然在形成水凝胶的过程中起到重要作用.本节中将分别讨论拓扑形状和超分子作用力在水凝胶形成中的作用.
3.1 形状和尺寸在GO水凝胶形成中的作用
研究表明,GO的大小对水凝胶的形成有重要的作用.在最早Luo等[12]所报道的GO自发形成的水凝胶中,GO是通过氧化膨胀石墨的方法制得的,其平均面积高达112 μm2,远大于Hummers法氧化天然石墨所得到的普通GO.大尺寸的GO在0.3%的质量分数下即自发形成水凝胶,这表明在该质量分数下,GO片在溶液中已经相互接触,形成三维网络聚集体.而普通GO以同样的质量分数(0.3%)分散于水中,只能形成溶液.显然,在相同质量分数时,大尺寸的GO片之间相互接触的面积更大,有利于形成三维网络.
Bai等[23]比较了2种大小不同的GO形成复合水凝胶的过程,发现GO的大小对水凝胶形成具有决定性的作用,如图3所示.他们通过使用不同大小的天然石墨作为原料,利用Hummers法制得了大尺寸GO(平均直径大于5 μm)和小尺寸GO(直径小于2 μm).实验结果表明,大尺寸GO在酸化时发生凝胶化,而小尺寸GO同样条件下酸化时生成了沉淀.作者认为,酸化时GO片在水中的稳定性下降,将发生聚集.由于层层堆积得到的结构在GO片层间存在多重氢键,所以是一种能量上更为有利的聚集体.小尺寸的GO回转半径较小,可以方便地调整自身的取向,因此倾向于形成堆积结构,从而在溶液中析出.而大尺寸的GO片回转半径很大,无法迅速调节构象和片的取向,聚集时基本保持了溶液中的随机取向,GO片之间相互支撑,形成三维网络.这一结果表明GO较大的平面尺寸有利于三维网络的形成.
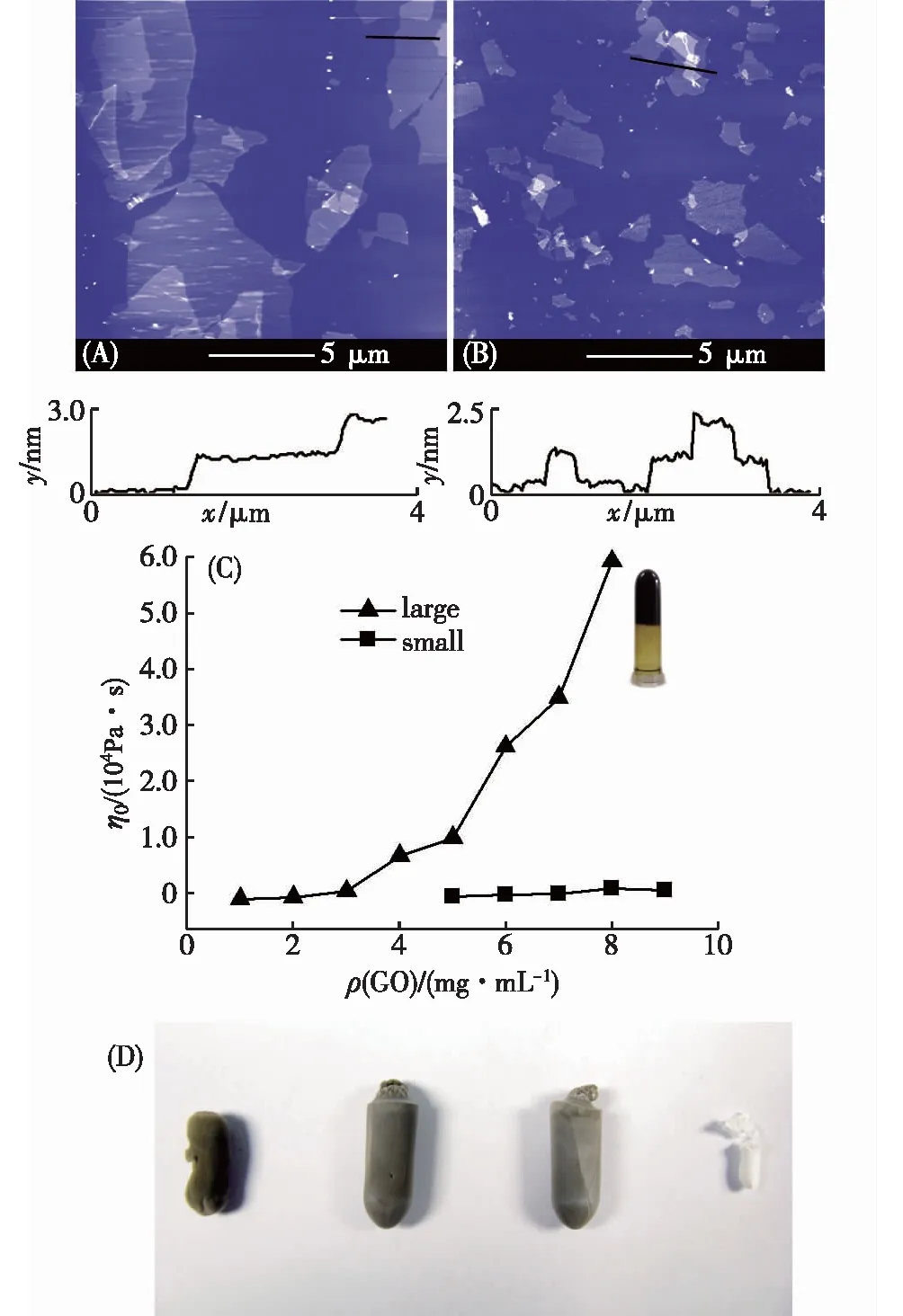
(A)大尺寸GO;(B)小尺寸GO;(C)上述2种 GO不同浓度的分散液在pH为0.6时的零切黏度, 插图为5 mg/mL的大尺寸GO分散液形成水凝胶的照片; (D)不同浓度的GO和PVA冷冻干燥后的照片, 从左到右依次为:1,3,5 mg/mL GO和3 mg/mL PVA.图3 GO尺寸大小对水凝胶形成的作用 [23]Fig.3 The influence of GO size on the form ation of hydrogel [23]
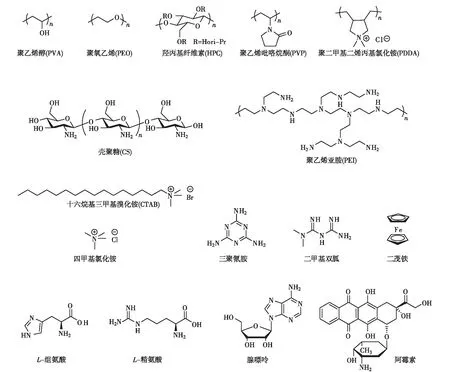
图4 用于制备GO水凝胶的典型的超分子交联剂Fig.4 Typical supramolecular crosslinking agents for the preparation of GO hydrogels
Bai等[23]还比较了二维的GO与一维的线性高分子之间的区别.他们发现通过冷冻干燥的方法处理0.3%的GO溶液,可以得到和初始溶液体积及形状均相同的GO疏松多孔固体.而处理同样浓度的PVA溶液,得到的固体样品体积远小于初始溶液.这表明在质量分数为0.3%时,二维GO片在溶液中已经相互接触,除去溶剂后能形成牢固的三维网络;而一维的PVA分子则无法在这一质量分数的溶液中形成三维网络.其原因在于GO片尺寸较大,在溶液中构象较为伸展,低质量分数时即可相互接触;此外,二维的GO片本身具有一定的强度,可以保证形成的网络的力学性能.而一维的聚合物在水中采取无规线团的构象,尺寸较小,未能形成亚浓溶液;且一维分子容易变形,只有多个分子形成聚集体之后才能有足够的强度,进一步增加了形成三维网络的临界浓度.因此,GO的二维形状在水凝胶形成过程中起到了重要作用.该作者进一步推测,GO在临界浓度以上的溶液中,形成了动态三维网络,其中结合力和排斥力达到平衡.当增强GO片之间的结合力,或者削弱GO片之间的排斥力时,动态网络被增强,溶液就会凝胶化.
3.2 GO水凝胶中的超分子作用力
为促进GO凝胶化,需调节GO片之间的作用力.上文已经提到,GO表面拥有丰富的含氧基团,可以提供多种超分子作用.因此,我们可以通过添加多种超分子交联剂,改变GO片之间的作用力平衡,诱导水凝胶的形成.常用于制备GO水凝胶的典型的超分子交联剂如图4所示.
3.2.1 氢键作用
氢键作用由GO上的各种含氧基团提供.GO上的含氧基团数目众多,与之形成多重氢键的物质能够显著增强GO之间的结合力.非离子型的水溶性高分子通常都带有含氧亲水基团,是氢键的给体或者受体,因此可以作为GO的氢键交联剂.Bai等[13]将GO和PVA溶液混合后振荡10 s并超声20 min,得到含有5 mg/mL GO和0.5~2.5 mg/mL PVA的GO/PVA的水凝胶,作者认为GO和PVA之间的多重氢键是水凝胶形成的驱动力.作者还研究了PVA交联剂浓度对凝胶过程的影响,发现凝胶过程很大程度上取决于PVA与GO的质量比:当GO的质量浓度在5 mg/mL时,只有在PVA与GO的质量比在1∶10~1∶2之间才能形成稳定的水凝胶,如图5所示.作者认为,当PVA质量浓度较低时,无法形成足够的交联密度;而PVA质量分数过高时,PVA将GO完全包裹,溶液整体性质类似于PVA溶液,同样不能形成水凝胶.

(A)图从左到右m(PVA)∶m(GO)=1∶1, 1∶1.5,1∶2,1∶5,1∶10,1∶20,1∶40.图5 GO/PVA复合物照片(A)及GO/PVA 复合水凝胶的pH响应性(B)[13]Fig.5 Photographs of GO/PVA mixtures(A), and pH dependent sol-gel transition of GO/PVA composite hydrogel (B) [13]
类似的,其他水溶性聚合物,如PEO、HPC、PVP,也可以作为氢键交联剂,诱导GO的凝胶化[23].PEO和PVP是氢键的受体,HPC不仅是受体而且还是供体,三者与GO都能形成氢键,提供额外的结合力,从而形成稳定的网络结构.实验结果还表明,水溶性聚合物的分子质量对形成水凝胶的力学性能有影响,分子质量较大的交联剂制得的水凝胶黏度更大.这是因为交联剂分子链长增大,则与2片GO所形成多重氢键的概率亦增大.同理,小分子氢键给受体往往不能起到交联作用.这是因为在水溶液中,氢键交联剂与GO之间的氢键面临着水分子氢键的竞争,而且氢键作用距离较短.所以只有能形成多重氢键且分子尺寸较大的聚合物才能同时与不同的GO片作用,起到交联效果.Bai等[23]证实HPC作为一种多糖衍生物可以通过氢键交联GO形成水凝胶,而小分子的葡萄糖则不能诱导GO凝胶化.
上述氢键交联的GO水凝胶都是对pH敏感的:提高水凝胶的pH值,可以导致GO上羧基的电离程度增加,斥力增大,水凝胶分解成溶液(如图5(B)所示)[13,23].如果选用合适的交联剂制备GO水凝胶,则还可以实现其他的环境响应性.Sahu等[24]在纳米尺寸的GO中加入少量的聚醚嵌段共聚物(聚氧乙烯-聚氧丙烯-聚氧乙烯,Pluronic),制备可注射型热敏性的水凝胶,可在接近人体体温时发生溶胶-凝胶转变.氢键在水凝胶形成过程中起到交联作用.此外,作者观察到了疏水的聚氧丙烯链段长度对水凝胶的形成有影响,所以作者认为除氢键作用外,疏水作用在水凝胶的形成中也起到了作用.
3.2.2 静电作用
GO上的电荷作用主要来自于羧基的电离,所以GO是一种带负电的粒子.负电荷之间的斥力是GO能在水中分散的原因[25].由于羧基是一种弱酸,其电离程度可以通过pH调节,因此,改变pH值可以削弱静电斥力,导致GO凝胶化.这一实验被Bai等[23]完成.他们发现,当酸化0.5%的GO溶液时,溶液的黏度急剧上升,形成水凝胶.ζ-电势的研究发现,酸化过程中GO的ζ-电势从-45 mV逐渐上升到0 mV左右.因此,酸化导致了GO电离程度下降,负电荷密度降低,静电斥力下降,发生聚集.这一结果证明了GO片之间的静电斥力在GO凝胶形成过程中起了很重要的作用.
更一般的,为了调节GO之间的静电作用,可以在溶液中添加带有正电荷的交联剂.Bai等[23]为了研究静电相互作用对GO凝胶化的影响,选择PDDA作为交联剂.由于PDDA没有能形成氢键的位点,所以使用PDDA作为交联剂可以排除氢键的干扰.实验表明PDDA上的季铵官能团与GO片上的羰基发生静电作用,在很低的浓度就可以促使GO的凝胶化.

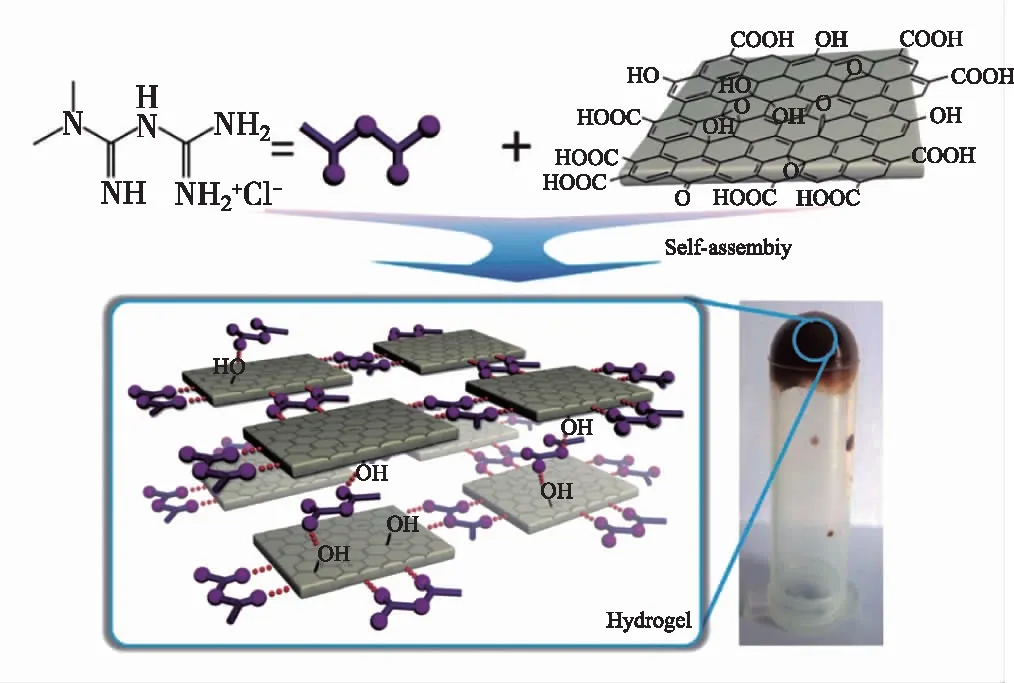
图6 MFH为交联剂的GO复合 水凝胶的照片以及形成过程示意图[28]Fig.6 GO composite hydrogel with MFH as crosslinking agent[28]
3.2.3 π-π作用
GO上残存的未氧化区域可以提供π-π作用.与上面所讨论的情况类似,如果在GO分散液中加入能与GO产生π-π作用的物质,则能够将GO交联形成水凝胶.Qiu等[31]报道了一个典型的例子,在这一工作中,他们使用GO作为分散剂,在水溶液中分散碳纳米管(CNT).由于CNT和GO的共轭区域之间存在很强的π-π作用,同时GO又有很强的亲水性,因此GO可以在水中有效地分散CNT.当CNT质量分数达到0.5%时,混合分散液发生了凝胶化,如图7所示.作者所使用的CNT是未经修饰的CNT,其他基团的含量极低.所以,上述水凝胶的形成主要是依靠π-π作用.在另一个工作中,Ai等[32]使用二茂铁作为交联剂,成功制得了GO水凝胶.由于二茂铁中不含有极性基团,所以作者认为二茂铁和GO之间的π-π作用是水凝胶形成的驱动力.

图7 GO和CNT形成的复合水凝胶的照片[31]Fig.7 Photograph of GO/CNT composite hydrogel[31]
事实上,利用纯粹的π-π作用制备GO水凝胶的方法较少见,这是因为,为了保证具有大π体系的交联剂在水中有一定的分散能力,交联剂通常会带有亲水基团,因此无法避免地引入氢键、静电作用等其他超分子作用.但是,在一些水凝胶体系中,π-π作用的确成为水凝胶交联的重要作用力.Xu等[33]将GO溶液与双链DNA溶液均匀混合后90 ℃下加热,使双链DNA解旋,利用单链DNA与相邻的GO之间的π-π作用,可以制得GO/DNA复合水凝胶.由于DNA中存在大量极性基团,所以不能排除氢键参与了凝胶的形成.但是Raman光谱的结果表明,GO和DNA之间存在电子转移,因此二者之间的π-π作用必定存在.同时,π-π作用受到pH影响很小,这也可以解释上述水凝胶为何能在强酸、强碱中1周后仍然保持稳定.
此外,Bai等[22]研究了CP在GO片分散液中进行原位化学聚合制备GO/CP复合水凝胶,如图8所示.作者认为GO片上共轭结构会与CP主链之间发生π-π作用,起到交联的效果.另外,氧化态CP与GO的静电作用,以及CP与GO之间可能的氢键作用,共同参与形成了GO/CP网络结构.上述水凝胶的储能模量接近105Pa,但是比较易碎,这可能是CP的高模量和低伸长率所导致的.

图8 GO/聚吡咯(PPy)复合水凝胶的照片 (A)和电子显微镜照片(B)[22]Fig.8 Photograph (A) and SEM image (B) of GO/polypyrrole composite hydrogel[22]
3.2.4 配位作用
配位作用来源于GO上的众多含氧基团.这些含氧基团可以和很多金属离子发生配位[34].Bai等[23]研究发现,向GO溶液中加入金属离子,包括Ca2+、Mg2+、Cu2+、Pb2+、Cr3+、Fe3+,会促进GO水凝胶的形成,但是K+、Li+、Ag+不能引起GO凝胶化.作者指出,多价金属离子和GO的配位作用是GO发生凝胶化的驱动力.为了证实这一点,作者在上述水凝胶中加入螯合剂乙二胺四乙酸(EDTA),发现水凝胶迅速转化为溶液.这是由于EDTA与金属的配位常数更大,将金属离子从GO表面夺走,水凝胶失去金属离子的交联作用,即发生溶液化.Huang等[35]也报道了La3+离子交联的GO水凝胶,同样,在水凝胶中加入EDTA可以发生凝胶-溶胶转变.
4 GO水凝胶的应用
水凝胶具有良好的生物相容性、负载性能、渗透性,在很多领域有重要的应用.GO水凝胶结合了GO独特二维形状、巨大比表面积的特点,所以具有一些传统水凝胶所不具有的特性.在本节中,我们将对GO自组装水凝胶在环境、生物医学等领域的应用做简要的总结.
4.1 吸附剂
近年来,随着工业迅速发展,环境污染日趋严重,水污染问题得到了越来越多的关注.污水中含有大量的有毒染料和重金属,这些污染对人体有严重的伤害,会导致营养不良、疾病甚至死亡[36-37],因此污水治理成为迫切的问题.吸附一直是处理污水的一个重要方法.由于GO自组装水凝胶具有很大的比表面积,GO片上有多种吸附位点,而水凝胶中联通的孔洞又有利于物质的扩散,所以GO自组装水凝胶很适合作为污染物吸附剂.
Xu等[33]研究了GO/DNA复合水凝胶对番红O染料的吸附.由于复合水凝胶大的比表面积,以及带负电的GO和DNA与带正电的番红O之间的强静电作用和π-π作用,复合水凝胶对番红O的吸附量可以达到960 mg/g.Chen等[21]设计合成了GO/CS复合水凝胶.由于GO与CS分别能够提供带正电和带负电的吸附位点,所以GO/CS复合水凝胶对带正电的亚甲基蓝(MB)和带负电的伊红(Eosin Y)吸附量都在300 mg/g以上.而其他常规吸附剂,如酸处理的活性炭,只能对其中一类染料有较高的吸附作用.此外,上述水凝胶对Cu(II)和Pb(II)的吸附量在测量范围内最大值分别是70 和90 mg/g.因此,GO/CS是一种广谱吸附剂,对水中常见的污染物,包括阴阳离子染料和金属离子,都具有良好的吸附能力.上述GO/CS复合水凝胶可以制成交换柱,使用过滤的方法处理含有染料的污水,如图9所示.
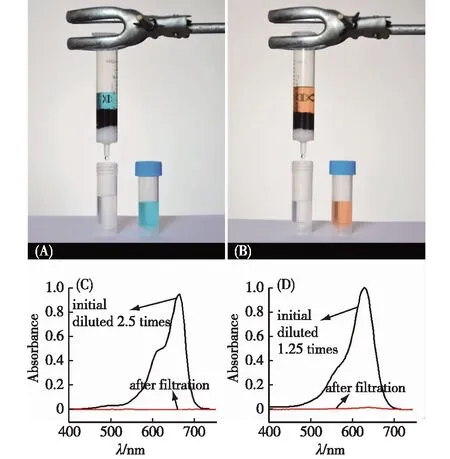
(A)吸附柱以及含有MB的溶液过滤前后的照片; (B)吸附栓以及含有Eosin Y的溶液过滤前后的照片; (C)过滤前后MB和溶液的吸收光谱; (D)过滤前后Eosin Y溶液的吸收光谱[21].图9 使用GO/CS复合水凝胶制成的 吸附柱处理含有染料的污水Fig.9 Removal of dyes from water by GO/CS composite hydrogel
除了用于在水中吸附污染物之外,GO水凝胶通过冷冻干燥的方法制得的气凝胶还可以用于吸附气体.Huang等[38]测得PEI和La3+交联的GO气凝胶对H2S的吸附量分别为46.7和63.5 mmol/g,两者的吸附量都比活性炭的吸附量(20 mmol/g)要高[39].作者还发现,对于其他一些还原性气体,如SO2,HI和H2S,上述气凝胶都有良好的吸附能力.由于上述吸附都是在室温常压下发生,因此作者认为,相比于活性炭吸附剂,上述GO气凝胶吸附剂具有明显的优势.
4.2 控制药物释放
水凝胶的一个重要应用即负载药物和控制药物释放.GO复合水凝胶具有环境响应性,同时GO有良好的生物相容性,所以GO水凝胶具有很好的药物释放能力.Bai等[13]研究了负载了维生素B12的GO/PVA水凝胶在不同pH值下的释放特性.作者发现,在中性磷酸盐缓冲液中,42 h后维生素B12释放率达到84%,而在酸溶液中释放率仅为51%.作者认为,在酸性条件下,GO/PVA复合水凝胶中GO之间的排斥力下降,结合更紧密,导致维生素B12的扩散路径增长.这种pH可调控的释放特性可以用于控制口服药物的释放,减少药物在酸性胃液中的释放,使其定向在肠道中释放,以避免药物对胃的刺激,或者药物在酸性条件下的分解.
4.3 气体传感器
CP具有良好的室温敏感性,能与很多气体作用,发生掺杂/去掺杂或者物理吸附,产生自身电导率的变化[40].为了提高CP气体传感器的灵敏度,CP层需要尽可能薄,以降低待测气体在聚合物中的扩散长度.Bai等[22]将合成的GO/PPy复合水凝胶冷冻干燥,得到多孔的GO/PPy气凝胶.由于在GO/PPy水凝胶中,PPy与GO的质量比接近1∶1,由此可以推断,PPy均匀分布在GO三维网络上,形成了一个非常薄的薄层.这种气凝胶作为电阻型传感器的敏感元件,对氨气的响应信号的强度远高于普通电化学聚合得到的薄膜,如图10所示.同时作者也发现,冷冻干燥得到的多孔结构对于提高传感器的灵敏度很重要.如果GO/PPy水凝胶在空气中自然干燥,则可得到致密的复合材料,这种材料对氨气响应很差.其原因在于氨气无法快速扩散进入致密的GO/PPy复合材料.
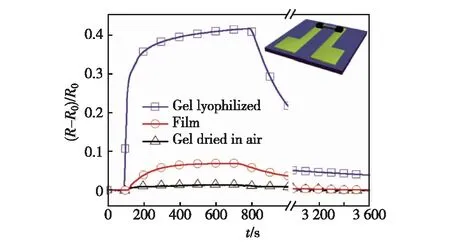
图10 传感器对氨气的传感性能[22]Fig.10 Ammonia gas sensing performance of sensor devices[22]
5 GO水凝胶的展望
综上,由于GO具有独特的二维拓扑形状,和丰富的表面基团,因此GO在水溶液中很容易与其他物质通过各种超分子相互作用进行自组装,形成水凝胶.GO水凝胶具有制备简便,性能多样化的特点,在环境、生物医药等领域有广阔的应用前景.在上述工作的基础上,我们认为,下列几点将是GO水凝胶领域今后所需着重研究解决的问题:1) GO水凝胶的可控合成.目前,尚未见到可以对GO水凝胶的各种性质进行精确调控的方法,这些性质与GO和交联剂之间的关系还没有完全明确.例如,凝胶—溶胶转变的pH值对于药物释放有重要的作用,但是合成具有指定临界pH的GO凝胶还比较困难.这些工作的完成,一方面依赖于GO原料的可控合成,另一方面依赖于我们对凝胶形成过程的更深入理解.2) GO水凝胶化学的基本理论.对于传统的高分子凝胶,已经有一系列理论可以描述水凝胶的微观结构与性能.对于GO水凝胶这种由二维材料构成的水凝胶,上述理论的实用性尚未得到验证.例如,GO水凝胶的流变性能、溶胀性能、溶质在GO水凝胶中的扩散过程等,都需要从理论上加以阐释.3) GO水凝胶的应用.虽然GO水凝胶已经取得了一些应用,但GO水凝胶的潜力并未完全得到发挥.因此,在今后的工作中,我们需要发展新的交联剂,赋予GO水凝胶新的功能,结合GO大比表面积和二维拓扑形状的特点,开发GO水凝胶更多的应用.相信在石墨烯材料高速发展的背景下,未来将有更多的GO水凝胶出现,这些新水凝胶必将能推动凝胶理论的完善,而且在越来越多的领域取得应用的突破.
[1] Geim A K.Graphene:status and prospects[J].Science,2009,324(5934):1530-1534.
[2] Novoselov K S,Fal′ko V I,Colombo L,et al.A roadmap for graphene[J].Nature,2012,490(7419):192-200.
[3] Stankovich S,Piner R D,Chen X Q,et al.Stable aqueous dispersions of graphitic nanoplatelets via the reduction of exfoliated graphite oxide in the presence of poly(sodium 4-styrenesulfonate)[J].Journal of Materials Chemistry,2006,16(2):155-158.
[4] Li D,Müller M B,Gilje S,et al.Processable aqueous dispersions of graphene nanosheets[J].Nature Nanotechnology,2008,3(2):101-105.
[5] Xu Y X,Bai H,Lu G W,et al.Flexible graphene films via the filtration of water-soluble noncovalent functionalized graphene sheets[J].J Am Chem Soc,2008,130(18):5856-5857.
[6] Pei S F,Cheng H M.The reduction of graphene oxide[J].Carbon,2012,50(9):3210-3228.
[7] Dreyer D R,Park S,Bielawski C W,et al.The chemistry of graphene oxide[J].Chemical Society Reviews,2010,39(1):228-240.
[8] Kim J E,Han T H,Lee S H,et al.Graphene oxide liquid crystals[J].Angew Chem Int Ed,2011,50(13):3043-3047.
[9] Ji L W,Rao M M,Zheng H M,et al.Graphene oxide as a sulfur immobilizer in high performance lithium/sulfur cells[J].J Am Chem Soc,2011,133(46):18522-18525.
[10] Loh K P,Bao Q L,Eda G,et al.Graphene oxide as a chemically tunable platform for optical applications[J].Nature Chemistry,2010,2(12):1015-1024.
[11] Dikin D A,Stankovich S,Zimney E J,et al.Preparation and characterization of graphene oxide paper[J].Nature,2007,448(7152):457-460.
[12] Luo Z T,Lu Y,Somers L A,et al.High yield preparation of macroscopic graphene oxide membranes[J].J Am Chem Soc,2009,131(3):898-899.
[13] Bai H,Li C,Wang X L,et al.A pH-sensitive graphene oxide composite hydrogel[J].Chemical Communications,2010,46(14):2376-2378.
[14] Hummers W S,Offeman R E.Preparation of graphitic oxide[J].J Am Chem Soc,1958,80(6):1339.
[15] Cai W W,Piner R D,Stadermann F J,et al.Synthesis and solid-state NMR structural characterization of13C-labeled graphite oxide[J].Science,2008,321(5897):1815-1817.
[16] Li Z Y,Zhang W H,Luo Y,et al.How graphene is cut upon oxidation?[J].J Am Chem Soc,2009,131(18):6320-6321.
[17] Brodie B C.On the atomic weight of graphite[J].Philosophical Transactions of the Royal Society,1859,149:249-259.
[18] Lerf A,He H Y,Forster M,et al.Structure of graphite oxide revisited[J].Journal of Physical Chemistry B,1998,102(23):4477-4482.
[19] Gao W,Alemany L B,Ci L J,et al.New insights into the structure and reduction of graphite oxide[J].Nature Chemistry,2009,1(5):403-408.
[20] Dimiev A,Kosynkin D V,Alemany L B,et al.Pristine graphite oxide[J].J Am Chem Soc,2012,134(5):2815-2822.
[21] Chen Y Q,Chen L B,Bai H,et al.Graphene oxide-chitosan composite hydrogels as broad-spectrum adsorbents for water purification[J].Journal of Materials Chemistry A,2013,1(6):1992-2001.
[22] Bai H,Sheng K X,Zhang P F,et al.Graphene oxide/conducting polymer composite hydrogels[J].Journal of Materials Chemistry,2011,21(46):18653-18658.
[23] Bai H,Wang X L,Li C,et al.On the gelation of graphene oxide[J].Journal of Physical Chemistry C,2011,115:5545-5551.
[24] Sahu A,Choi W I,Tae G.A stimuli-sensitive injectable graphene oxide composite hydrogel[J].Chemical Communications,2012,48(47):5820-5822.
[25] Wang X L,Bai H,Shi G Q.Size fractionation of graphene oxide sheets by pH-assisted selective sedimentation[J].J Am Chem Soc,2011,133(16):6338-6342.
[26] Huang C C,Bai H,Li C,et al.A graphene oxide/hemoglobin composite hydrogel for enzymatic catalysis in organic solvents[J].Chemical Communications,2011,47(17):4962-4964.
[27] Yang Q,Wang Z B,Weng J.Self-assembly of natural tripeptide glutathione triggered by graphene oxide[J].Soft Matter,2012,8(38):9855-9863.
[28] Tao C A,Wang J F,Qin S Q,et al.Fabrication of pH-sensitive graphene oxide-drug supramolecular hydrogels as controlled release systems[J].Journal of Materials Chemistry,2012,22(47):24856-24861.
[29] Adhikari B,Biswas A,Banerjee A.Graphene oxide-based hydrogels to make metal nanoparticle-containing reduced graphene oxide-based functional hybrid hydrogels[J].ACS Applied Materials & Interfaces,2012,4(10):5472-5482.
[30] Adhikari B,Biswas A,Banerjee A.Graphene oxide-based supramolecular hydrogels for making nanohybrid systems with Au nanoparticles[J].Langmuir,2012,28(2):1460-1469.
[31] Qiu L,Yang X W,Gou X L,et al.Dispersing carbon nanotubes with graphene oxide in water and synergistic effects between graphene derivatives[J].Chemistry A European Journal,2010,16(35):10653-10658.
[32] Ai W,Du Z Z,Liu J Q,et al.Formation of graphene oxide gel via the π-stacked supramolecular self-assembly[J].RSC Advances,2012,2(32):12204-12209.
[33] Xu Y X,Wu Q,Sun Y Q,et al.Three-dimensional self-assembly of graphene oxide and DNA into multifunctional hydrogels[J].ACS Nano,2010,4(12):7358-7362.
[34] Park S,Lee K S,Bozoklu G,et al.Graphene oxide papers modified by divalent ions-enhancing mechanical properties via chemical cross-linking[J].ACS Nano,2008,2(3):572-578.
[35] Huang H,Lu S Y,Zhang X T,et al.Glucono-δ-lactone controlled assembly of graphene oxide hydrogels with selectively reversible gel-sol transition[J].Soft Matter,2012,8(17):4609-4615.
[36] Shannon M A,Bohn P W,Elimelech M,et al.Science and technology for water purification in the coming decades[J].Nature,2008,452(7185):301-310.
[37] Montgomery M A,Elimelech M.Water and sanitation in developing countries including health in the equation[J].Environmental Science & Technology,2007,41:17-24.
[38] Huang H,Chen P W,Zhang X T,et al.Edge-to-edge assembled graphene oxide aerogels with outstanding mechanical performance and superhigh chemical activity[J].Small,2013,9(8):1397-1404.
[39] Bagreev A,Bandosz T J.H2S adsorption/oxidation on unmodified activated carbons:importance of prehumidification[J].Carbon,2001,39:2303-2311.
[40] Bai H,Shi G Q.Gas sensors based on conducting polymers[J].Sensors,2007,7:267-307.
猜你喜欢
高中数理化(2022年14期)2022-08-15
辽宁化工(2022年5期)2022-05-28
波谱学杂志(2021年3期)2021-09-07
粘接(2021年2期)2021-06-10
原子与分子物理学报(2019年5期)2019-04-28
铜仁学院学报(2018年6期)2018-07-05
橡胶科技(2016年2期)2016-07-30
中学化学(2015年12期)2016-01-19
橡塑技术与装备(2015年7期)2015-07-03
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
