
Catalytic Cracking of Cycloparaffins Admixed with Olefins: 2. Single-Event Microkinetic (SEMK) Assessment
2014-07-25 10:07:52XueGaopingWengHuixinJorisThybautGuyMarin
中国炼油与石油化工 2014年2期
Xue Gaoping; Weng Huixin; Joris W. Thybaut; Guy B. Marin
(1 Research Institute of Petroleum Processing, East China University of Science and Technology, Shanghai 200237, China; 2. Laboratory for Chemical Technology, Ghent University, Ghent, B-9000, Belgium)
Catalytic Cracking of Cycloparaffins Admixed with Olefins: 2. Single-Event Microkinetic (SEMK) Assessment
Xue Gaoping1,2; Weng Huixin1; Joris W. Thybaut2; Guy B. Marin2
(1 Research Institute of Petroleum Processing, East China University of Science and Technology, Shanghai 200237, China; 2. Laboratory for Chemical Technology, Ghent University, Ghent, B-9000, Belgium)
The developed SEMK model is used to provide an insight into the contribution of individual reactions in the cracking of methylcyclohexane as well as the site coverage by various carbenium ions. The preferred reaction pathways for the conversion of methylcyclohexane are hydride transfer reactions followed by PCP-isomerizations, deprotonation and endocyclic β-scission, accounting for 61%, 22% and 12% of its disappearance, respectively, at 693 K and 30% conversion of methylcyclohexane. Protolysis plays a minor role in the cracking of methylcyclohexane. Once cyclic diolefins are formed, all of them can be instantaneously transformed to aromatics, which are easily interconverted via disproportionation. Judging from the carbenium ion concentrations it is evident that, at the investigated operating conditions, less than 5% of the acid sites are covered by carbenium ions, less than 2% of which corresponds to cyclic type species including allylic ones.
catalytic cracking; single-event microkinetic model; cycloparaffin; olefin; site coverage by carbenium ions
1 Introduction
From the 1990s on, developments in analytical techniques and computer science have led to kinetically modeling complex reaction systems at the molecular level (microkinetic model). Catalytic cracking, as a typical complex reaction system, has been drawing extensive attention of researchers from all over the world. Structure model[1-2], structure-oriented model[3], single event model[4]and the model employing linear free energy relationship[5-6]are the most effective ways to adequately address the complex product distribution of catalytic cracking over a wide range of operating conditions.
Up to date, the single-event methodology, structureoriented model and the model employing linear free energy relationship are still actively developing, while there are few reports on the further development of the structure model. The former three types of microkinetic models utilize different properties of the elementary reactions to simulate catalytic cracking process from different perspectives. The ultimate goal of these models is to reasonably correlate the proposed mechanism with the experimental observations. The predictions provided by the models are generally consistent with the experimental measurements and the models are accordingly considered as successful. However, there were few studies on the further exploration of the microkinetic models. Lots of instructive information could be extracted from these reliable kinetic models,e.g., contribution analysis and acid site coverage by carbenium ions, which can allow one to better understand the underlying cracking chemistry.
In our first paper[7], the Single-Event MicroKinetic (SEMK) model for the catalytic cracking of methylcyclohexane admixed with 1-octene was effectively enhanced and the predictions about cyclic C6and C8species have been significantly improved. In this paper, we use this enhanced SEMK model to quantitatively analyze the reactions that occur during catalytic cracking and address the role of different reactions that primarily determine the activity and selectivity. The acid site coverage by various carbenium ions is also presented which is a valuable reference for quantum chemical calculations of catalysis. To match with the non-equilibrium between olefins and alkylcarbenium ions, the (de)protonation affinities between cyclic hydrocarbons and the corresponding cyclic carbe-nium ions are calculated and the non-equilibrium between the latter is further confirmed.
2 Experimental and Validation of the SEMK model
Experiments have been performed previously on a commercial REUSY zeolite in a TEOM®microbalance reactor[8]. Methylcyclohexane was used as the feedstock admixed with 17.65 mol% of 1-octene to initiate feedstock conversion more easily. The reaction temperature ranged from 693 K to 753 K where thermal cracking was confirmed to play a negligible role in the cracking system.
A space-time between 12.09 and 42.30 kgcat·s/mol was applied to cover a broad range of methylcyclohexane conversion rates,i.e., between 9.7% and 54.5%. Under these reaction conditions, the absence of transport limitations in the pores of the catalyst has been con firmed. More experimental details can be found elsewhere[8].
Going through reaction network generation, relumping scheme and parameter estimation, the SEMK model for the catalytic cracking of cycloparaffins has been established[7]. Then the model is validated through comparison of the calculated yields of measurable species with the experimental ones together with the physical analysis of the obtained parameters,i. e., activation energies. From the discussion section of our first paper[7], it can be seen that the enhanced SEMK model can adequately capture the cracking behavior of cycloparaf fins with conversion and temperature. The enhanced SEMK model is therefore considered as reasonable and reliable.
3 Discussion
By making use of the reliable SEMK model, the following information can be explored and extracted. These information not only allows one to grasp the underlying catalytic cracking chemistry and analyze the roles of different reactions in the whole reaction pattern, but also sheds some light on the phenomena, which the present experimental techniques cannot delve into.
3.1 Contribution analysis
The relative importance of the various reactions in the cracking system can be assessed by a contribution analysis based on the obtained activation energies and the employed operating conditions. Such an analysis allows for a better understanding of the reaction mechanism and a more clear recognition of the prevailing reaction pathways.
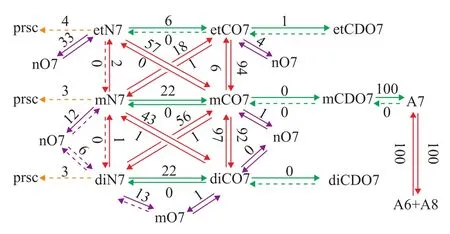
Figure 1 Reaction network of the catalytic cracking of methylcyclohexane admixed with 1-octene
Figure 1 presents the corresponding analysis at 753 K and 30% of methylcyclohexane conversion obtained using the enhanced SEMK model. With respect to the feed,i.e., methylcyclohexane (mN7), exocyclic protolysis can directly occur resulting in methane and cyclohexa(e)ne. Hydride transfer reactions of mN7 followed by endocyclic β-scissions can produce normal heptene (nO7) which can subsequently break down into lower carbon-number hydrocarbon fragments. Methylcyclohexene (mCO7) can be formed from mN7 by hydride transfer reactions followed by deprotonation. Ethylcyclopentane (etN7), dimethylcyclopentane (diN7), ethylcyclopentene (etCO7) and dimethylcyclopentene (diCO7) can be produced from mN7 via hydride transfer reactions followed by PCP-isomerization. It should be noted that hydride transfer reactions are the prerequisite for mN7 to produce nO7, mCO7, etN7, diN7, etCO7 and diCO7, so its reaction rate can significantly affect or even control the production rate of the latter species. It can be seen from Figure 1 that the preferred reaction pathways for conversion of the feed are hydride transfer reactions followed either by PCP-isomerization and deprotonation or solely by deprotonation to cyclic olefins, accounting for 61% and 22% of its disappearance, respectively. 12% of the feed is cracked via endocyclic β-scission to nO7 which will yield smallerparaf fins and ole fins, while protolysis plays a minor role in cracking methylcyclohexane which only accounts for 3% of its conversion but constitutes the main pathway towards C6cyclic hydrocarbons.
The methylcyclohexane isomers,i.e., etN7 and diN7 are mainly formed via PCP-isomerization from mCO7 rather than directly from mN7, as can be determined from the thickness of the arrows in Figure 1. Remarkably, the reverse transformation of these isomers back to mCO7 is predominant in their disappearance, i.e., 57% of etN7 and 56% of diN7 are converted back to mCO7 via PCP-isomerization. The cracking of etN7 and diN7 follows the same behavior as that of mN7. Endocyclic β-scission contributes 33% and 13% to the ring opening of etN7 and diN7, respectively, while 3%—4% of etN7 and diN7 are cracked via protolysis.
Cyclic ole fins,i.e., etCO7, mCO7 and diCO7 are mainly formed from mN7 via hydride transfer followed by PCP-isomerization and deprotonation. The PCP-isomerization between these cyclic olefins are the fastest reactions in the cracking of methylcyclohexane at the investigated operating conditions. Although the relative contribution of endocyclic β-scission to the consumption of the cyclic olefins is only minor,which accounts for4% at its maximum, and in absolute terms a signi ficant amount of cyclic olefins undergoes ring-opening reaction, resulting in a considerable formation of acyclic hydrocarbons, such as nO7 and methylhexene (mO7), which then would break down to smaller hydrocarbons, e.g. propa(e)ne and buta(e)ne. Hence, it can be concluded that except for the direct cracking of mN7, smaller acyclic hydrocarbons are substantially formed from the ring-opening cracking of its cyclic ole finic isomers.
Cyclic diolefins,i.e., ethylcyclopentadiene (etCDO7), methylcyclohexadiene (mCDO7) and dimethylcyclopentadiene (diCDO7), are exclusively formed from their corresponding cyclic ole finic counterparts via hydride transfer followed by deprotonation involving allylic species. Sincelargely exceedsthe rate of formation of cyclic diolefins from cyclic olefins is much higher than that of the rate of transformation of cyclic diole fins to cyclic ole fins. Similarly to the ring opening of cyclic ole fins, the formation of cyclic diole fins constitutes a substantial amount but only a minor fraction of the cyclic ole fins.
With respect to aromatics, toluene (A7) is the key intermediate. About all mCDO7 species are transformed to A7 via hydride transfer followed by deprotonation involving allylic species. Due to the considerably high activation energy for hydride transfer reactions consuming allylic carbenium ions, even though the aromatic ring can be easily protonated to cyclic diolefinic carbenium ions, and the latter is marginally converted back to cyclic diolefins via hydride transfer reactions. As a result, practically no aromatics (0%) are converted to cyclic diolefins, and further to cyclic olefins, leading to ring opening reaction of aromatics. Instead, all A7 species are transformed via fast disproportionation to benzene (A6) and xylene (A8), which in turn are converted back to A7, resulting in formation of other aromatics, such as trimethylbenzene (A9) which is not shown in Figure 1.
The temperature effect on the contribution analysis of the cracking of methylcyclohexane is mainly associated with the contribution of protolysis through hydride transfer. Lower temperature (693 K) results in lower contribution of protolysis (1%) to the methylcyclohexane transformation at a 30% conversion. At lower methylcyclohexane conversion (1%) and higher temperature (753 K), only 4% of methylcyclohexane is converted via protolysis, and hydride transfer primarily accounts for the activation of methylcyclohexane. For the case ofn-decane and 1-octene under the same operating conditions, the protolysis initially plays a key role in then-decane conversion even when it is admixed with olefins. It can be attributed to the significantly lower hydride transfer of methylcyclohexane (122.0—127.8 kJ/mol) than that ofn-decane (135.5 kJ/mol). Based on the above-mentioned study on the contribution analysis of the cracking of methylcyclohexane, it can be clearly seen that cyclic olefins play a quite active role in the conversion of mN7. Its naphthenic isomers are mainly produced from mCO7 rather than directly from mN7. Besides the direct cracking of mN7, the cracking of its cyclic olefinic isomers significantly contributes to the formation of products from ring-opening reaction. The aromatization of cycloparaffins to aromatics can be considered irreversible based on the comparison of rates of the forward and backward reactions involved. Aromatics are primarily interconverted to each other via facile disproportionation.It is notable that some of the aboveobservations extracted from the contribution analysis of the cracking of methylcycohexane admixed with 1-octene in this work are significantly different from one presented by Quintana-Solorzano,et al.[9]First of all, Quintana-Solorzano,et al.[9]concluded that the hydride transfer followed by deprotonation between mN7 and mCO7 was the fastest reaction in the cracking system, while the PCP-isomerization between mCO7 and etCO7 as well as that between mCO7 and diCO7 was considered the fastest in this work. Moreover, etN7 and diN7 were significantly formed from mN7 in Quintana-Solorzano’s work, whereas the former two were primarily formed from mCO7 rather than from mN7. Secondly, unlike the conclusion drawn from Quintana-Solorzano’s work that only cyclic olefins,i.e., etCO7, mCO7 and diCO7, would undergo ring-opening reaction to form acyclics, it is shown in this work that the endocyclic β-scission of cyclic hydrocarbons, i.e., etN7, mN7 and diN7, also significantly contributes to the formation of acyclics. Thirdly, it was observed by Quintana-Solorzano that 25% of A7 would be transformed to mCDO7 via protonation followed by hydride transfer and the disproportionation between aromatics, i.e., the interconversion of A7 to A6 and A8, proceeds at a reaction rate comparable to the rate of the transformation of A7 to mCDO7. However, it is shown in this work that almost none of A7 transforms to mCDO7 and all the A7 can be interconvertible to A6 and A8 via fast disproportionation.
3.2 Acid site coverage
Intermediates play a key role in the catalytic cracking of hydrocarbons over zeolites. The discussion regarding carbenium ions versus alkoxy species as the real intermediates in catalytic cracking over zeolites has been ongoing for years and still remains a subject of debate[10]. However, the existence of heavier cyclic carbenium ions in zeolite pores,e. g., 1,3-dimethylcyclopentenyl cation[11], benzenium cation[12]and 1,2,4,5-tetramethylbenzenium cation[13]has been experimentally confirmed. Even though smaller alkyl carbenium ions, such ast-butyl cation, have not been experimentally observed, several quantum chemical calculations concerning isobutene protonation in zeolite with differing pore sizes revealed that the zeolite framework can increase the stability of carbenium ions compared to alkoxy species so as to facilitate the formation of the former instead of the latter[10,14-15]. The SEMK model considers carbenium ions as the intermediates and carbenium ion-like species as the transition state involved in the elementary steps[4].
3.2.1 Total carbenium ion concentration
The total concentration of carbenium ions is obtained by multiplying the concentration of the acid sites and the acid site coverage by all carbenium ions. The concentration of the acid sites is estimated to be 1.83×10-4kmol/kg by the regression of experimental data[16]. When the acid site coverage by carbenium ions was calculated, a pseudo steady-state approximation could be made. The concentration of carbenium ions is expected to be very low and remains a constant with time. So their disappearance rate should be equal to their formation rate. Here, only adsorption and desorption reactions are considered. For instance, an alkylcarbenium ion can be formed either by protonation of an olefin molecule, or by hydride transfer of a paraffin molecule, or by protolysis of a large paraffin molecule. Meanwhile, it can be consumed either by deprotonation, or by hydride transfer with an arbitrary paraffin. By summing up the disappearance rates and formation rates of all carbenium ions, an expression could be obtained which involves hydrocarbon partial pressures, number of single events and the single-event rate coefficients. With regard to the single event rate coefficients, the activation energies for cyclics[7]and those obtained by Gaoping,et al.[17]for acyclics together with the preexponential factors calculated by Quintana-Solorzano,et al.[16]have been used. Figure 2 shows the correspondingly calculated total acid site coverage as a function of the methylcyclohexane conversion at various reaction temperatures for the catalytic cracking of methylcyclohexane and 1-octene.
Under the conditions investigated, less than 5% of the total active sites are covered by carbenium ions, which is in accordance with the results obtained by Yaluris,et al.[6]and Van Borm,et al.[18]At a given reaction temperature, the evolution of the acid site coverage with conversion is complicated by the admixture of 1-octene. Initially before the methylcyclohexane conversion reaches 4%, the acid site coverage by carbenium ions sharply increases with the conversion as a result of the easily occurringprotonation of 1-octene. This phenomenon is evidenced by the relatively low activation energy of 111.8 kJ/mol for protonation of olefins[17]compared to 142.1—161.3 kJ/mol which is required for the protolysis of cycloparaffins. Subsequent cracking of the octyl carbenium ions results in smaller carbenium ions that can occupy the free active sites of the catalyst. Upon depletion of 1-octene and, hence, slowing down in supplying the fast carbenium ions, small carbenium ions have to be produced from methylcyclohexane via hydride transfer followed by ring opening reactions and consequent β-scission. With an activation energy in the range of 122.0—127.8 kJ/mol this type of hydride transfer is significantly slower than octene protonation and, hence, the surface coverage by carbenium ions decreases. Also the low concentration of carbenium ions on the surface contributes to the lower rate of the bimolecular hydride transfer compared to the protonation, which only requires an available acid site. However, at higher methylcyclohexane conversion more methylcyclohexyl carbenium ions have been produced, and since dimethylcyclopentyl and ethylcyclopentyl carbenium ions are susceptible to ring opening reaction, the concentration of carbenium ions again increases and even rises beyond the maximum value obtained at lower conversions. At a given conversion level, the acid site coverage is significantly lower at higher temperatures. As a matter of fact, the acid site coverage is mainly determined by the (de)protonation behavior of olefinic species with the temperature. Olefins can be protonated to alkylcarbenium ions and the latter can be deprotonated to the former. The similar relationship exists for conversion of cyclic olefins to cyclic carbenium ions, and for transformation of cyclic diolefins or aromatics to allylic carbenium ions. So the acid site coverage by all the carbenium ions is dictated by this (de)protonation relationship.
Deprotonation activation energies required for all types of carbenium ions generally exceed those required for the corresponding protonation reactions. For example, for the case of (de)protonation between olefins and alkylcarbenium ions the deprotonation activation energy is 178.4—219.2 kJ/mol, while the olefin protonation activation energy is 93.7—111.8 kJ/mol[17]. With regard to cyclic carbenium ions and cyclic olefins, the deprotonation energy is 178.5—228.2 kJ/mol, while the cyclic olefins protonation energy is in the range of 121.8—122.3 kJ/mol. For protonation of cyclic diolefins or aromatics to allylic carbenium ions, the deprotonation activation energy is 179.0—200.3 kJ/mol, while the protonation activation energy is 95.0—110.6 kJ/mol.
As a result, deprotonation is enhanced more remarkably by higher temperatures than protonation does, resulting in a tendency for carbenium ions to be desorbed as olefins, cyclic olefins, cyclic diolefins or aromatics in the gas phase, which would ultimately lead to lower acid site coverage at higher temperature. Correspondingly, the maximum in the carbenium ion concentration at lower methylcyclohexane conversion is most pronounced at lower temperatures[17].

Figure 2 The evolution of acid site coverage as a function of conversion at various temperatures for the catalytic cracking of methylcyclohexane and 1-octene
3.2.2 Surface concentration of various carbenium ion types
Table 1 shows the concentration of detailed carbenium ion types during methylcyclohexane conversion which is equal to 1% and 10% at reaction temperature of 693 K and 753 K, respectively. About 98%—99% of the carbenium ions adsorbed on the catalyst surface correspond to acyclic tertiary carbenium ions. Acyclic secondary carbenium ions and all cyclic carbenium ions only account for less than 2% of the covered acid sites. This is in line with the highest chemisorption heat of 125.5 kJ/mol that is obtained for olefin protonation towards tertiary alkyl carbenium ions, as shown in Table 2. Among the latter, tertiary pentyl and butyl carbenium ions,i.e., the smallest ones, are predominant. They are likely to be formed from 1-octene by β-scission. The cracking of methylcyclohex-ane also significantly contributes to the formation of t-butyl carbenium ions after hydride transfer, ring opening reaction and β-scission. Likewise, among the secondary acyclic carbenium ions, propyl and secondary butyl carbenium ions that are the smallest secondary carbenium ions are prevailing. This is in line with the experimental observation that propa(e)ne and buta(e)ne are the main acyclic products in the reactor effluents. Among the cyclic carbenium ions, tertiary methylcyclohexyl carbenium ions are more abundant than secondary ones. The enthalpy of cyclic olefins protonation towards tertiary cyclic carbenium ions is equal to 105.9 kJ/mol and is much higher than that yielding secondary ones, i.e., 56.7 kJ/mol. As it is expected, allylic cyclic carbenium ions are generally more stable than simple cyclic carbenium ions since the corresponding protonation enthalpies are estimated to be 89.7 kJ/mol and 56.7 kJ/mol, respectively. Unlike the general trend that tertiary carbenium ions are more stable than secondary ones, the concentration of tertiary allylic cyclic carbenium ions is similar to that of secondary ones, as evidenced by similar protonation enthalpies already reported in Table 2.

Table 1 Acid site coverage by typical carbenium ions at 10% methylcyclohexane conversion and 753K

Table 2 Reaction enthalpy for protonation of olefins, cyclic olefins, cyclic diolefins and aromatics yielding the corresponding alkyl, cyclic, and allylic cyclic carbenium ionskJ/mol
It can be seen from the above-mentioned investigation of the acid site coverage of various carbenium ions that similar to the case of the cracking of paraffins and olefins[17]the tertiary acyclic carbenium ions account for most of the intermediates adsorbed on the catalyst surface. Tertiary butyl and pentyl carbenium ions are prevailing since they are the smallest ions which cannot undergo further cracking reactions. Acyclic secondary and cyclic carbenium ions, in particular the allylic ones, only account for less than 2% of the covered acid sites.
3.3 Non-equilibrium between cyclic hydrocarbons and cyclic carbenium ions
It is noticed that analogous to the non-equilibrium between olefins and alkyl carbenium ions in the cracking ofn-decane and 1-octene[17], the (de)protonation of cyclic olefins, cyclic diolefins and aromatics to form cyclic and allylic cyclic carbenium ions, respectively, are not in equilibrium under the conditions investigated thereby. At 693 K and a methylcyclohexane conversion ranging from 1% to 30%, the (de)protonation affinity for methylcyclohexene to secondary and tertiary methylcyclohexyl carbenium ions spans in the range from -26 307 to -21 193 J/mol and from 8 541 to 20 999 J/mol, respectively. For protonation of methylcyclohexadiene and toluene, the (de)protonation affinity also significantly exceeds 2 000 J/mol, indicating that (de) protonation involving both conventional cyclic and allylic cyclic carbenium ions have not reached equilibrium as compared to those involving conventional alkyl carbenium ions[17].
4 Conclusions
It can be recapitulated from the reliable SEMK model that under the reaction conditions investigated thereby methylcyclohexane is mainly consumed via hydride transfer followed by PCP-isomerization, deprotonation and en-docyclic β-scission. Protolysis plays a minor role in the cracking of methylcyclohexane. The cyclic olefinic isomers of methylcyclohexane are the main intermediates involved in the transformation of the latter to its naphthenic isomers. Endocyclic β-scission reactions can significantly convert methylcyclohexane and its cyclic olefinic isomers to form products after ring-opening reaction. The aromatization of cycloparaffins to form aromatics can be considered irreversible and the formed aromatics are primarily interconverted to each other via disproportionation. Less than 5% of the active sites of the catalyst are occupied by carbenium ions. Among the various carbenium ions adsorbed on the catalyst, 98%—99% are of tertiary acyclic type in which t-pentyl and t-butyl carbenium ions are dominant. Other carbenium ions including cyclic and allylic species only account for less than 2% of the carbenium ions adsorbed. Among these cyclic and allylic carbenium ions, tertiary cyclic carbenium ions are more stable than secondary ones and allylic carbenium ions are generally more stable than cyclic ones.
Acknowledgements:The authors acknowledge the financial support from the China Scholarship Council and the Long Term Structural Methusalem Funding by the Flemish Government.
Reference
[1] Liguras D K, Allen D T. Structural models for catalytic cracking. 1. Model-compound reactions[J]. Ind Eng Chem Res, 1989, 28(6): 665-673
[2] Liguras D K, Allen D T. Structural models for catalytic cracking. 2. Reactions of simulated oil mixtures [J]. Ind Eng Chem Res, 1989, 28(6): 674-683
[3] Quann R J, Jaffe S B. Structure-Oriented Lumping: Describing the Chemistry of Complex Hydrocarbon Mixtures[J]. Ind Eng Chem Res, 1992, 31(11): 2483-2497
[4] Feng W, Vynckier E, Froment G F. Single-event kinetics of catalytic cracking[J]. Ind Eng Chem Res, 1993, 32(12): 2997-3005
[5] Yaluris G, Rekoske J E, Aparicio L M, et al. Isobutane cracking over Y-zeolites: I. Development of a kineticmodel[J]. J Catal, 1995, 153(1): 54-64
[6] Yaluris G, Rekoske J E, Aparicio L M, et al. Isobutane cracking over Y-zeolites: II. Catalytic cycles and reaction selectivity[J]. J Catal, 1995, 153(1): 65-75
[7] Xue Gaoping, Weng Huixin, Thyabut J W, et al. Catalytic cracking of cycloparaffins admixed with olefins: 1. Singleevent microkinetic (SEMK) modeling[J]. China Petroleum Processing & Petrochemical Technology, 2014, 16(1): 71-80
[8] Quintana-Solorzano R, Thybaut J W, Marin G B. Catalytic cracking and coking of (cyclo)alkane/1-octene mixtures on an equilibrium catalyst[J]. Appl Catal A: General, 2006, 314(2): 184-199
[9] Quintana-Solorzano R. Single-event microkinetics for coking in catalytic cracking: Development and application[D]. Ghent: Ghent University, 2007
[10] Nguyen C M, De Moor B A, Reyniers M F, et al. Isobutene protonation in H-Fau, H-Mor, H-ZSM-5, and H-ZSM-22[J]. J Phys Chem C, 2012, 116(34): 18236-18249
[11] Chua Y T, Stair P C, Nicholas J B, et al. UV Raman spectrum of 1,3-dimethylcyclopentenyl cation adsorbed in zeolite H-MFI[J]. J Am Chem Soc, 2003, 125(4): 866-867
[12] Bjorgen M, Bonino F, Kolboe S, et al. Spectroscopic evidence for a persistent benzenium cation in zeolite HBeta[J]. J Am Chem Soc, 2003, 125(51): 15863-15868
[13] Bjorgen M, Bonino F, Arstad B, et al. Persistent methylbenzenium ions in protonated zeolites: The required proton affinity of the guest hydrocarbon[J]. Chem Phys Chem, 2005, 6(2): 232-235
[14] Boronat M, Zicovich-Wilson C M, Viruela P, et al. Cluster and periodic calculations of the ethene protonation reaction catalyzed by theta-1 zeolite: Influence of method, model size, and structural constraints[J]. Chemistry-A European Journal, 2001, 7(6): 1295-1303
[15] Fang H J, Zheng a M, Li S H, et al. New insights into the effects of acid strength on the solid acid-catalyzed reaction: Theoretical calculation study of olefinic hydrocarbon protonation reaction[J]. J Phys Chem C, 2010, 114(22): 10254-10264
[16] Quintana-Solorzano R, Thybaut J W, Marin G B. A singleevent microkinetic analysis of the catalytic cracking of (cyclo) alkanes on an equilibrium catalyst in the absence of coke formation[J]. Chem Eng Sci, 2007, 62(18/20): 5033-5038
[17] Xue Gaoping, Thyabut J W, Weng Huixin, et al. Singleevent microkinetic (SEMK) assessment of the catalytic cracking of alkanes admixed with alkenes[J]. Submitted to Ind Eng Chem Res
[18] Van Borm R, Reyniers M-F, Marin G B. Catalytic cracking of alkanes on FAU: Single-event microkinetic modeling including acidity descriptors[J]. AIChE Journal, 2012, 58(7): 2202-2215
Recieved date: 2013-10-22; Accepted date: 2013-12-30.
Weng Huixin, Telephone: +86-21-64252816; E-mail: hxweng@ecust.edu.cn.

- 中国炼油与石油化工的其它文章
- Synthesis of PE with Broad MWD Catalyzed by Supported Ziegler-Natta Catalyst Consisting of Cycloalkoxy Silane as IED
- Deep Extractive Desulfurization of Gasoline with Ionic Liquids Based on Metal Halide
- Synthesis of Macro-Mesostructured γ-Al2O3with Large Pore Volume and High Surface Area by a Facile Secondary Reforming Method
- Performance of FCC Catalyst Improved with Vanadium Trapping Components
- Synthesis, Characterization and Evaluation of Sulfur Transfer Catalysts for FCC Flue Gas
- Selection of Chelated Fe (III)/Fe (II) Catalytic Oxidation Agents for Desulfurization Based on Iron Complexation Method