
GPCR A2AAR Agonist Binding and Induced Conformation Changes of Functional Switches
2014-07-19 11:17:08XueqinPangJianyongLiu
Xue-qin Pang,Jian-yong Liu
State Key Laboratory of Molecular Reaction Dynamics,Dalian Institute of Chemical Physics,Chinese Academy of Sciences,Dalian 116023,China
GPCR A2AAR Agonist Binding and Induced Conformation Changes of Functional Switches
Xue-qin Pang,Jian-yong Liu∗
State Key Laboratory of Molecular Reaction Dynamics,Dalian Institute of Chemical Physics,Chinese Academy of Sciences,Dalian 116023,China
Agonist binding of A2Aadenosine receptor(A2AAR)shows protective e ff ects against infl ammatory and immune.E ff orts are exerted in understanding the general mechanism and developing A2AAR selectively binding agonists.Using molecular dynamics(MD)simulations,we have studied the interactions between A2AAR and its agonist(adenosine),and analyzed the induced dynamic behaviors of the receptor.Key residues interacting with adenosine are identi fied:A632.61,I662.64,V843.32,L853.33,T883.36,F1685.29,M1775.38,L2496.51, H2506.52,and N2536.55interacting with adenosine with affinities larger than 0.5 kcal/mol. Moreover,no interaction between adenosine and L1675.28is observed,which supports our previous fi ndings that L1675.28is an antagonist speci fi c binding reside.The dynamic behaviors of agonist bound A2AAR are found to be di ff erent from apo-A2AAR in three typical functional switches:(i)tight“ionic lock”forms in adenosine-A2AAR,but it is in equilibrium between formation and breakage in apo-A2AAR;(ii)the“rotamer toggle switch”, T883.36/F2426.44/W2466.48,adopted di ff erent rotameric conformations in adenosine-A2AAR and apo-A2AAR;(iii)adenosine-A2AAR has a fl exible intracellular loop 2(IC2)and α-helical IC3,while apo-A2AAR preferred α-helical IC2 and fl exible IC3.Our results indicate that agonist binding induced di ff erent conformational rearrangements of these characteristic functional switches in adenosine-A2AAR and apo-A2AAR.
A2Aadenosine receptor,Molecular dynamics,Adenosine,Speci fi c binding, Conformational dynamics,Ionic lock,Rotamer toggle switch,Secondary structure
I.INTRODUCTION
Adenosine is produced under conditions of infection, injury,or stress.It acts as an endogenous agonist to A2Aadenosine receptor(A2AAR),which belongs to the G-protein coupled receptors(GPCRs)trans-membrane protein family.A2AAR adopts active and inactive allosteric equilibrium upon extracellular chemical stimuli[1].The binding of adenosine to A2AAR can activate the receptor and exert neural functions in the central nervous system[2],most possibly by elevating the intracellular level of cyclic AMP(cAMP)[3].It has been demonstrated that A2AAR agonist can suppress inf l ammation.Meanwhile,it has been observed in A2AAR silenced mice enhanced inf l ammatory response. Thus,agonist bound A2AAR shows protective effects against inf l ammatory and immune[4-6].In order to understand how agonist bound A2AAR suppresses the inf l ammatory responses,different mechanisms were proposed.The activation of NF-κB(nuclear factor kappalight-chain-enhancer of activated B cells)pathway was demonstrated f i rstly[7].But other studies revealed different pathways where the NF-κB was not activated[8, 9].The general mechanism of A2AAR activation is far from clear.
In the past decades,new achievements were obtained on determining the molecular structures and dynamic properties of A2AAR.Crystal structures binding with either agonist[10,11]or antagonist[12-14]were reported.These new structures lead to crucial hints to the functional mechanisms.Meanwhile,computational studies revealed dynamic information of A2AAR,and provided detailed understandings of its functionality. Conformational dynamics of A2AAR bound with agonist,adenosine or UK432097,suggested that the binding of adenosine was highly dynamical while UK432097 stabilized a much tighter neighborhood of active conformation.It thus explained the 100-to 1000-fold greater efficacy of UK432097 compared to adenosine [15].Simulations of A2AAR in both cholesterolfree and cholesterol-bound POPC(1-palmitoyl-2-oleoylsn-glycero-3-phosphocholine)membrane bilayers suggested that cholesterol could bind with A2AAR at the interface of TM1,TM2,and TM3,and be functioned as a cofactor with the agonist in GPCR activation[16].However,due to crystal packing and the low resolution of crystal structures(ranging from 2.6˚A to 3.6˚A),some loops of the protein are incomplete or shown in poor resolution.In this work,we describe the agonist binding at molecular level and illustrate important A2AAR conformation changes,which were not reported in previous studies on A2AAR agonist binding.
Adenosine is an endogenous agonist to A2AAR.It bounds to A2AAR and activates the receptor to exert neural functions in the central nervous system in human body[2].Drugs,either agonist or antagonist, will compete with adenosine for binding with A2AAR and then enhance or inhibit the activation of A2AAR, respectively.Thus to study the binding of adenosine and the induced dynamic behaviors of the receptor is of fundamental importance.This could be useful for the future experimental studies to unravel the molecular mechanism of A2AAR functioning.A2AAR shares the conserved topology structural fold in the GPCR protein family,in which the conformation of the highly conserved motifs and secondary structures of the loops rearrange,switching on and of fthe receptor.Among them,the“ionic lock”,“rotamer toggle switch”,and IC2/IC3 are identif i ed to be the characteristic functional switches of proteins in GPCR family.The conformations of these switches are different between active and inactive GPCRs[17-28].Fluorescence resonance energy transfer(FRET)[29]and site-directed spin labeling(SDSL)[30]could monitor the conformation adjustments of some key regions.However,direct tracking of the coherent conformation changes is difficult to achieve.
In this work,we studied the agonist binding and the induced conformational dynamics of the functional switches in A2AAR by comparing the conformations between apo-A2AAR and adenosine-A2AAR,and suggested structural indicators for A2AAR agonism.According to our observation,the ionic lock,rotamer toggle switch and secondary structures of IC2 and IC3, give different conformations between apo-A2AAR and adenosine-A2AAR.
II.METHODS
A.Structure preparation of A2AAR
The missing intracellular loop 3(IC3)and extracellular loop 2(EC2)were constructed by homo-modeling with Swiss Model[31].The vasopressin V2 receptor (PDBID 2JX4)and β2AR(PDB ID:2RH1)were used as templates,respectively.The generated structures were examined by Verify3D[32]and proven to be physical.Furthermore,a 1000-step energy minimization was performed by AMBER10 package[33].The H++ [34]was applied to determine the protonation state for titratable groups of the protein at pH=7.0.Missing hydrogen atoms in crystal structure were added by LEaP [33].
B.Construction of apo-A2AAR and adenosine-A2AAR
The apo-A2AAR model was obtained by removing the binding antagonist ZM241385 from the crystal structure.Adenosine-A2AAR model was prepared with Autodock 4.0 package[35]with the same protocol used in our previous work[36].The number of torsions in adenosine is 5.Referring to Autodock tutorial[35]and the reported applications[37],we performed 250 docking trials with 2.5×106poses evaluated in each trial and the one with the highest binding affinity was recorded in each trial.To cluster the 250 poses reported at the end of each docking experiment,we have tested the RMSD-tolerance of 2.0,1.0,and 0.5˚A.The RMSD-tolerance of 2.0˚A will cluster the binding modes into one big group and cannot classify different docking modes well. The RMSD-tolerance of 0.5˚A will cluster the binding modes into too many small groups.The RMSD-tolerance of 1.0˚A can cluster the binding modes into several representative groups.Thus,the resulting binding modes were clustered by RMSD-tolerance of 1.0˚A. Four biggest clusters with more than 15 members were gathered.To get the most probable binding mode,the one with lowest binding energy score in each of the four clusters was chosen to perform further MD simulation. The binding modes were further determined with MD simulations and detailed binding energy calculations.
C.System setup and force f i eld parameters for MD simulations
In VMD[38],each A2AAR model was inserted into a 100˚A×100˚A POPC bilayers.POPC molecules within 5.0˚A of any A2AAR atoms were removed.The A2AAR-lipid was solvated by 50˚A thick water layers in both sides.The full system was neutralized with Clcounter-ions with Amber.The f i nal system size was 100˚A×100˚A×159˚A,containing~1.32×105atoms.
MD simulations were performed by NAMD package [39]with Amber force f i eld FF99SB for the protein and GAFF for adenosine and POPC lipids.Atomic charges of adenosine were derived from R.E.D[40],in which structure optimizations and electrostatic potential calculations were taken with Gaussian 03[41]at the level of HF/6-31G∗.The partial charges were fitted using RESP algorithm.The atom types were allocated by ANTECHAMBER module.The atom charge and atom type of POPC was referred to that in Ref.[42].
D.Protocol of MD simulation
Firstly,the system was relaxed by a combined energy minimization and MD simulation scheme.The protein,adenosine,POPC molecules and crystal water were f i xed with a force constant of 2 kcal/(mol·˚A2),and the solvent was relaxed using 104-step energy minimization followed by 5×105-step NPγT MD simulation.Af-ter that,the constrain on POPC molecules was removed and the whole system was relaxed by another 104-step energy minimization and 5×105-step NPγT MD simulation.The agonist,crystal water and protein molecules were relaxed gradually using the same protocol.
The whole system was heated gradually to 310 K by Langevin dynamics with a damping coefficient of 1.0 ps-1.NPγT ensemble was used with the surface tension of 60 mN/m and the pressure of 1.01325 bar. The Langevin piston Nos´e-Hoover method was used to control the pressure[43,44],with the damping and oscillation time scales of Langevin piston as 50 and 100 fs, respectively.Covalent bonds involving hydrogen were restrained by the SHAKE algorithm[45].The shortranged nonbonded interaction was switched of fgradually from 10˚A to 12˚A.The PME method[46]was applied to treat long-range electrostatic interactions,and the grid size is 120˚A×120˚A×180˚A.A multiple-timestep algorithm was used with the covalent force evaluated every 1.0 fs.For the f i rst 10 ns simulation,the multiple time step scheme of 1.0 fs for short-range nonbonded force and 2.0 fs for long-range electrostatic force was used;then the time steps of 2.0 fs for short-range nonbonded force and 4.0 fs for long-range electrostatic force were used in the following simulation.
E.Binding energy decomposition
Binding energies between adenosine and protein molecules were calculated and decomposed on each residue of the protein with the MM-GBSA module in Amber[33].The calculations were performed according to the thermodynamic cycle shown in Fig.1.The energy terms were averaged over 103frames,which were extracted from each 10 ns trajectory in every 40th ns of the MD simulation.Snapshots of 40-50,90-100,and 140-150 ns were calculated,respectively.In the energy decomposition,the contributions of each term were decomposed to each residue of the protein.The binding mode selection of adenosine was performed in the same way.Since we mainly focused on which residues contribute greatly to ligand binding,we did not consider entropy contribution in this work[47-50].
F.Analysis protocols
The root-mean-square deviation(RMSD),distances between specif i c atom pairs,dihedral angles of residues, and hydrogen bonding interactions were analyzed by PTRAJ module of AMBER10.RMSD of protein and ligands were used to evaluate the convergence of the simulation.DSSP algorithm[51]was applied to assign the secondary structure content of IC2 and IC3 during MD simulation.
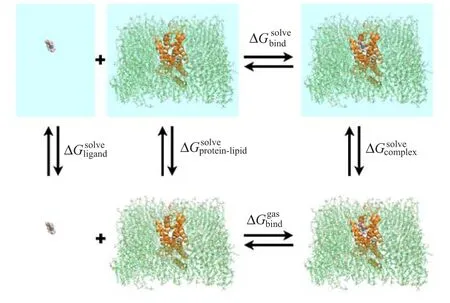
FIG.1 Thermodynamic cycle used in the binding free energy calculations.
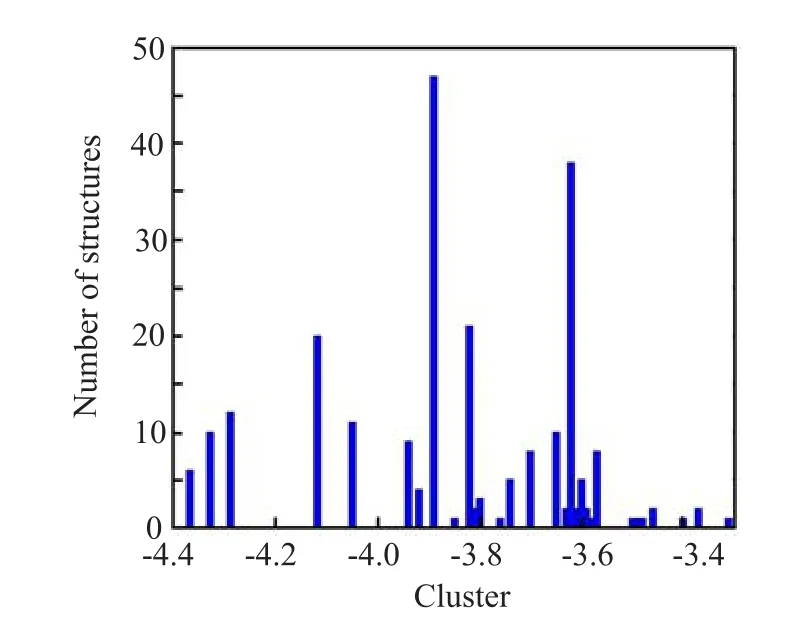
FIG.2 Clustered histogram of different binding poses for adenosine-A2AAR.RMSD-tolerance of 1.0˚A was used.
III.RESULTS AND DISCUSSION
A.Determined adenosine binding mode via docking and binding energy decomposition
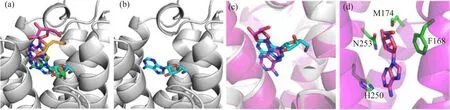
FIG.3 Adenosine-A2AAR binding mode illustrations.(a)Four selected binding modes predicted by Autodock.The rigid protein in docking process is shown in grey cartoon.Adenosine is shown in green,cyan,magenta and yellow sticks in the binding modes of Ade-4,Ade-8,Ade-10,and Ade-19,respectively.(b)Binding mode of Ade-8 predicted by Autodock. (c)Comparison between the Autodock result and the f i nal snapshot from short MD simulation of the Ade-8 binding mode. Ade-A2AAR from MD simulation is shown in cartoon and colored in magenta.(d)Final snapshot from the short MD simulation of Ade-8.The protein is shown in magenta cartoon,adenosine is shown in magenta sticks,and residues within 3 ˚A of adenosine are shown in green sticks.
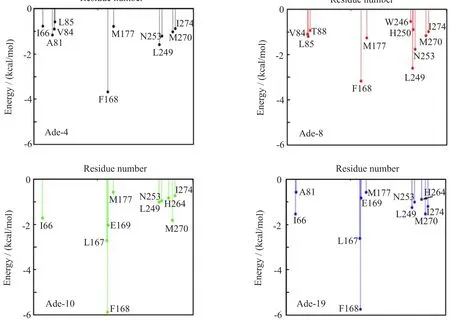
FIG.4 Binding energy decompositions of the four selected binding modes between adenosine and A2AAR.The total binding energy was decomposed on each residue of A2AAR and interactions greater than 0.5 kcal/mol were illustrated.
To determine the binding site of adenosine,we f i rst predicted the binding mode between adenosine and A2AAR by AutoDock,and then selected the most typical binding mode from short MD simulations and binding energy decomposition calculations.Four clusters with more than 15 members were gathered(Fig.2).To get the most probable binding mode,the one with lowest binding energy score in each of the 4 clusters were chosen for further calculations(Fig.2 and Fig.3(a,b)). Since the docking program did not consider the f l exibility of the protein structure,we performed MD simulations on the four most representative structures to achieve the equilibrium(Fig.3(c,d)).Binding energies between adenosine and protein were calculated with MM-GBSA in every 10 ps during the simulation, and further decomposed into terms of contributions on a per-residue basis for each binding mode.Residues, such as F1825.43,H2506.52,N2536.55,I2747.39,H2787.43, and S2817.46were reported to be important for agonist binding in experiments[52].Meanwhile,mutations of N2536.55A and H2506.52A would abolish agonist binding[14].It thus reveals that N2536.55and H2506.52play crucial roles in agonist binding.(Residues are labeled according to the Ballesteros-Weinstein residue numbering method[53],and supplementary material). As shown in Fig.4,in the binding mode of Ade-8,adenosine interacted strongly with most of those residues, especially with N2536.55and H2506.52.However,in the other three predicted binding modes,no interaction between adenosine and H2506.52,which is crucial for agonist binding,was detected.Therefore,we chose Ade-8 as the most reasonable binding mode for further MD simulation.Besides,all the interactions predicted in Ade-8 are observed in the recently reported agonist bound A2AAR crystal structure(PDBID 2YDO,but in poor resolution,3.0˚A),which proves the validity of our model[11].For the interaction observed in the crystal structure,all of the four binding modes failed to predict the interactions between adenosine and A632.61, N1815.42,and S2777.42.This might be because the f i xed protein did not equilibrate enough during the short MD simulation and the f l exibility of the protein was not fully considered during the binding energy calculation.However,it could also be an artifact of the poor resolution of the crystal structure.All of the predicted bindingmodes,as well as the recently published crystal structure,lack strong interactions between adenosine and F1825.43,H2787.43and S2817.46.Since the ligand binding experiments were done with other agonist rather than adenosine,different ligand types might be the reason for this.
B.Structure stability evaluation during MD simulation
The stability of the simulation was evaluated by RMSD with the crystal structure as refe˚rence.Rapidrises of the protein RMSD pro fi le(3-4A)within the fi rst 3 ns simulation suggested that protein conformation has been rearranged from the crystal structure(Fig.5(a)).These structure changes mainly resulted from two factors,one was the membrane environment,which indicated the di ff erence between the protein conformations in membrane(simulation)and in water(crystal);the other one was the replacement of exogenous T4L section(crystal)with homology-modeled structures of EC2 and IC3(simulation).The RMSD plateau after 20 ns suggested that the resulted structures were reasonably stable.As revealed by the fl uctuation of the adenosine RMSD(Fig.5(a)),the binding of adenosine is quite dynamic.Adenosine alternated its binding pose after~40 ns during the MD simulation, relative to the predicted binding mode.The adenosine in the new binding pose showed a similar conformation as the recent published adenosine-A2AAR crystal structure[11].However,in the MD simulations which started from the adenosine-A2AAR crystal,the bound adenosine also inverted compared to the crystal structure,and had similar conformation as we predicted[15]. Thus,our simulation also supported that adenosine was highly dynamic when it bound to A2AAR[15].
C.Identif i ed key adenosine-binding residues in A2AAR via interaction energy analysis
In order to identify important A2AAR residues that interacted with adenosine,we analyzed the interactions between A2AAR and adenosine by averaging the binding energy over 3 trajectories for adenosine-A2AAR. Residues with interactions stronger than 0.5 kcal/mol are shown in Fig.6.The detailed decompositions with standard deviation are shown in Table I.Among the experimentally reported residues,which are important to agonist binding[14,52],adenosine interacted strongly with H2506.52and N2536.55.Besides,the interactions between adenosine and A632.61,I662.64,V843.32, L853.33,T883.36,F1685.29,M1775.38and L2496.51were also detected.These interactions are consistent with the recently reported agonist bound A2AAR crystal structure,which proves the validity of our model[11]. In our previous work,L1675.28was found to be the antagonist specif i c binding residue,which only interactedwith antagonist but not agonist[36].During the MD simulation of adenosine-A2AAR,no strong interaction (binding affinity smaller than 0.2 kcal/mol)between L1675.28and adenosine was formed,which further supports our point.
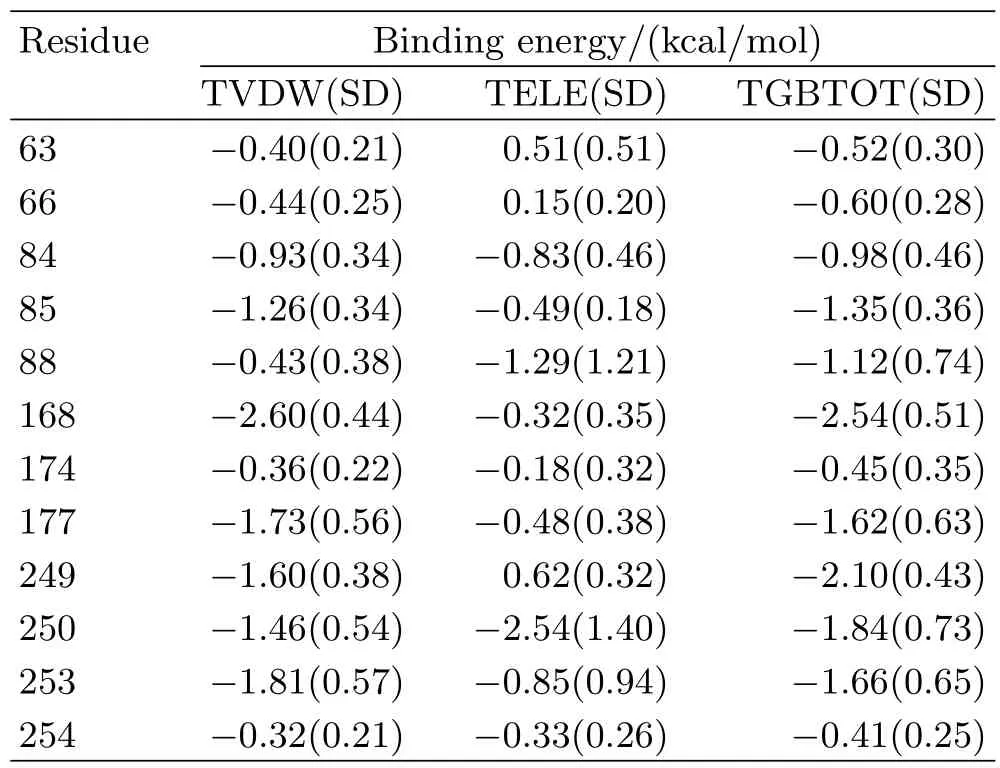
TABLE IBinding energy decomposition of adenosine-A2AAR in MD simulation.Residues,whose interactions with adenosine were stronger than 0.5 kcal/mol,are shown.
D.Adenosine binding induced the formation of the tight ionic-lock
The ionic lock was believed to be important in the inactive state of GPCR[14,17,23,54,55],but it was broken in the crystal structure of the inactive A2AAR [14].Our previous work suggested that the ionic lock in apo-A2AAR is equilibrated between forms of formation and that of breakage,while it stayed broken in the two antagonist binding holo-A2AARs[36].Besides,the analysis on hydrogen bond interactions of R1023.50and E2286.30suggested that the ionic lock is consisted of R1023.50-E2286.30,R1073.55-E2286.30,or R1023.50-H2306.32.In adenosine-A2AAR,the distances between R1023.50-E2286.30are the shortest.Thus we considered the R1023.50-E2286.30as the ionic lock.Figure 7 shows the time series of the shortest distance between this residue pair.In adenosine-A2AAR,salt bridge between R1023.50and E2286.30formed and maintained throughout the MD simulation.While in apo-A2AAR,the salt bridge between R1023.50and E2286.30formed and maintained within the f i rst 160 ns.Then the salt bridge between R1073.55and E2286.30formed from 190 ns to 200 ns.Compared to apo-A2AAR,for which the ionic lock equilibrated between lock and unlock states,adenosine-A2AAR formed tight ionic lock.The smaller ionic lock distances in adenosine-A2AAR indicated that agonist binding enhanced the lock and reduced the relative distance between TM3 and TM6.
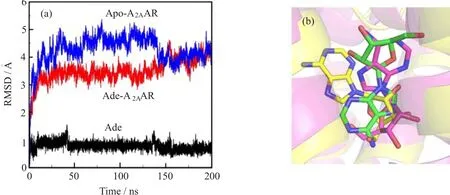
FIG.5(a)The RMSD of protein and adenosine during the MD simulation.Backbone atoms of the protein and heavy atoms of the bound ligand were included in the RMSD calculations.(b)Binding mode comparison between the simulation and recent published crystal structure.Adenosine structure is shown in sticks,with the predicted binding mode colored in green,the simulated binding mode at 50 ns in yellow and the crystal structure(PDBID:2ydo)in magenta.The protein at 50 ns is shown in yellow cartoon,while the 2ydo crystal structure is shown in magenta cartoon.
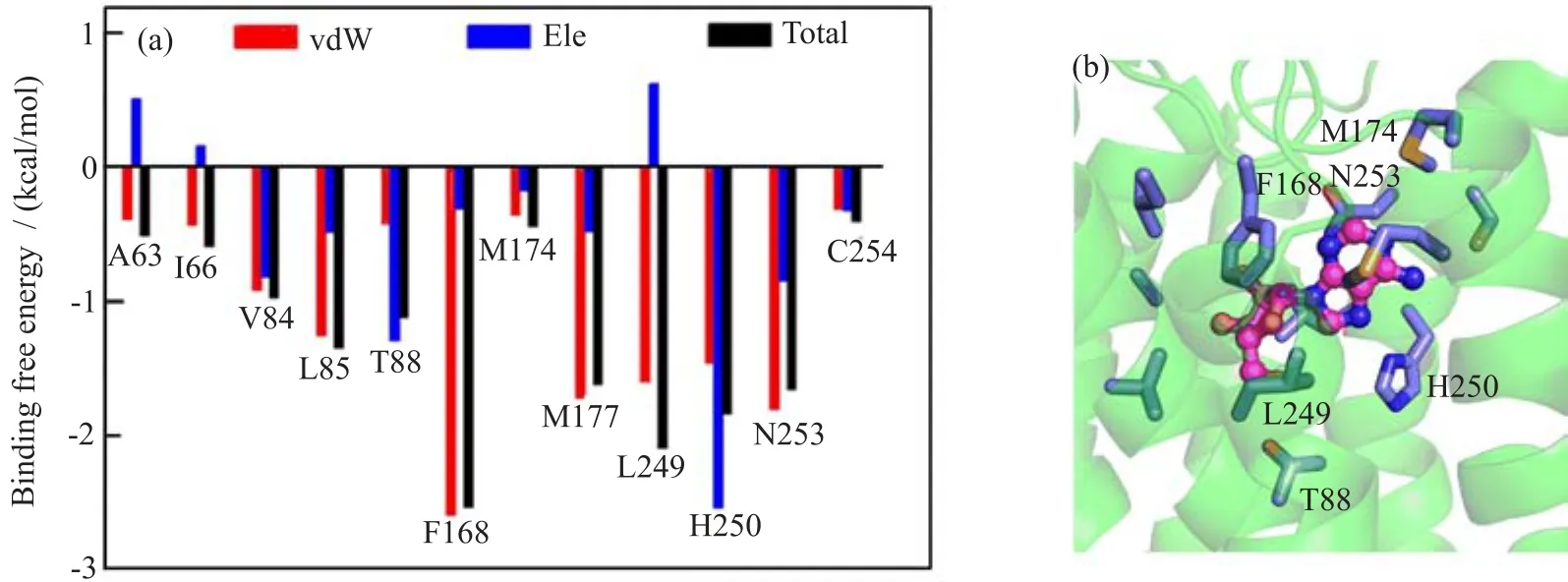
FIG.6 Protein-ligand interactions of adenosine-A2AAR.(a)Binding energy decomposition.(b)Illustration of interaction in adenosine-A2AAR.A2AAR is shown in green cartoon,adenosine is shown in magenta stick-spheres,residues interacted with adenosine are shown in blue sticks.
E.Adenosine binding induced rotamer toggle switch in A2AAR
Rotamer toggle switch was another structural element that could switch the GPCRs between active and inactive states by changing their χ1 rotamers(measured by the N-Cα-Cβ-Cγ torsion angle)in response to ligand binding[22].T883.36/F2426.44/W2466.48was identif i ed as the rotamer toggle switch in A2AAR.But this rotamer toggle switch adopted different rotation states between apo-A2AAR and adenosine-A2AAR.W2466.48remained in gauche-state(χ1 near-60◦)in adenosine-A2AAR,but shifted to trans(χ1 near±180◦)in apo-A2AAR(Fig.8).T883.36and F2426.44took trans rotamers in adenosine-A2AAR,but both of them switched to gauche-in apo-A2AAR(Fig.8).Rotamer toggle switch of the same conformations in adenosine-A2AAR is also reported in antagonist bound holo-A2AARs[36].
F.Antagonist binding induced secondary structure changes of IC2 and IC3

FIG.7 Ionic lock time series of apo-A2AAR and adenosine-A2AAR,which were assessed by both the side chain and backbone distances between residues involved.The corresponding distances observed in two inactive rhodopsin crystal structures (PDBID 1U19 and 1L9H)are indicated by orange lines.(a)The ionic lock of apo-A2AAR was evaluated by the closest side chain and backbone distances between residue pairs of R1023.50-E2286.30and R1073.55-E2286.30.The nearest N-O distance between R1023.50and E2286.30is shown in cyan,and the nearest N-O distance between R1073.55and E2286.30is shown in light pink.The smoothed side chain distances(N-O distances)and backbone distances(Cα-Cα distance) are shown in black and blue,respectively.(b)The ionic lock of adenosine-A2AAR was assessed by the distances between R1023.50and E2286.30.The distance between R102(NH1)and E228(OE1)is shown in light pink;the distance between R102(NH1)and E228(OE2)is shown in cyan,the nearest N-O distance is shown in black,while the Cα-Cα distance is shown in blue.
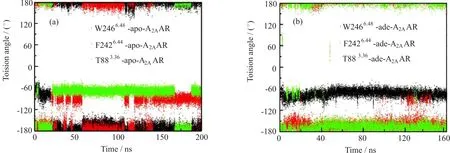
FIG.8 Rotamer changes of T883.36/F2426.44/W2466.48in apo-A2AAR(a)and adenosine-A2AAR(b).(gauche+,χ1 near 60◦;gauche-,χ1 near-60◦;and trans,χ1 near±180◦).W2466.48switched to a trans conformation in apo-A2AAR,whereas it remained gauche-in adenosine-A2AAR.F2426.44sampled frequently as gauche-in apo-A2AAR,whereas it remained trans in adenosine-A2AAR.T883.36frequently sampled gauche-in apo-A2AAR,whereas it mostly adopted trans conformations in adenosine-A2AAR.
For all GPCRs,IC2(TM3-IC2-TM4)and IC3(TM5-IC3-TM6)were believed to be“switch regions”that could alter the equilibrium between active and inactive states.The change in secondary structures of IC2 and IC3 could indicate the activation or inactivation of GPCR.The formation of α-helical IC2 might restrain GPCRs in their inactive states and weaken their binding to G proteins[24,28].The α-helical IC3 was suggested to be crucial for interactions between GPCR and the G proteins,and to further activate G proteins[56, 57].Our previous study showed that apo-A2AAR preferred α-helical IC2 and f l exible IC3;whereas in antagonist bound holo-A2AARs,an irregular IC2 and a short α-helical or 310-helical IC3 were more frequently observed[36].We compared the secondary structures of IC2 and IC3 between the apo-A2AAR and adenosine-A2AAR.In apo-A2AAR,IC2 adopted α-helix,while in adenosine-A2AARs it frequently behaved as irregular loops(Fig.9(a,c)and cartoons shown in Fig.10).For IC3,though the initial structure from homo-model was a long α-helix,it quickly transited to irregular loops and remained f l exible in apo-A2AAR,whereas a short α-helix(or 310-helix)was formed in adenosine-A2AAR (Fig.9(b),(d)and Fig.10).Thus,adenosine binding in A2AAR induced the secondary structure adjustments in IC2 and IC3,and the induced conformational changes are similar to those in antagonist bound A2AAR.The conformational changes of IC2 and IC3 in A2AAR couldbe a dynamic indicator for the binding of both agonist and antagonist.
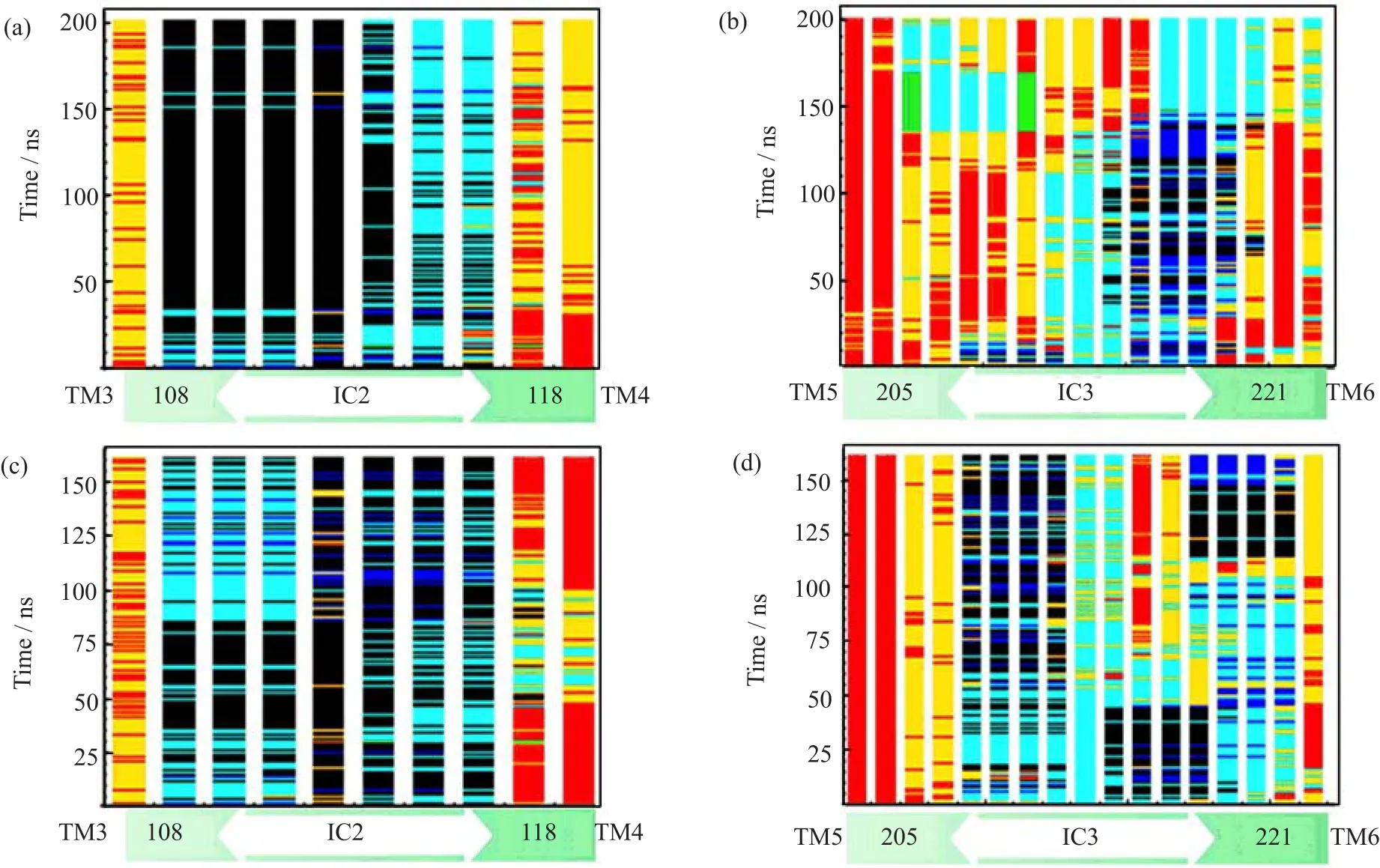
FIG.9 Secondary structure contents of IC2 and IC3 in the apo-A2AAR and adenosine-A2AAR during MD simulations. α-helix is colored in black,310-helix is colored in blue,extended strand in β ladder and isolated β-bridge are colored in green,hydrogen bonded turns,bends and coils are called loops,and colored in cyan,yellow and red,respectively.(a)IC2 in apo-A2AAR preferred a short α-helix.(b)IC3 adopted f l exible irregular loops in apo-A2AAR.(c)In adenosine-A2AAR, IC2 frequently exhibited irregular loops.(d)IC3 formed a short α-helix or 310-helix in adenosine-A2AAR.
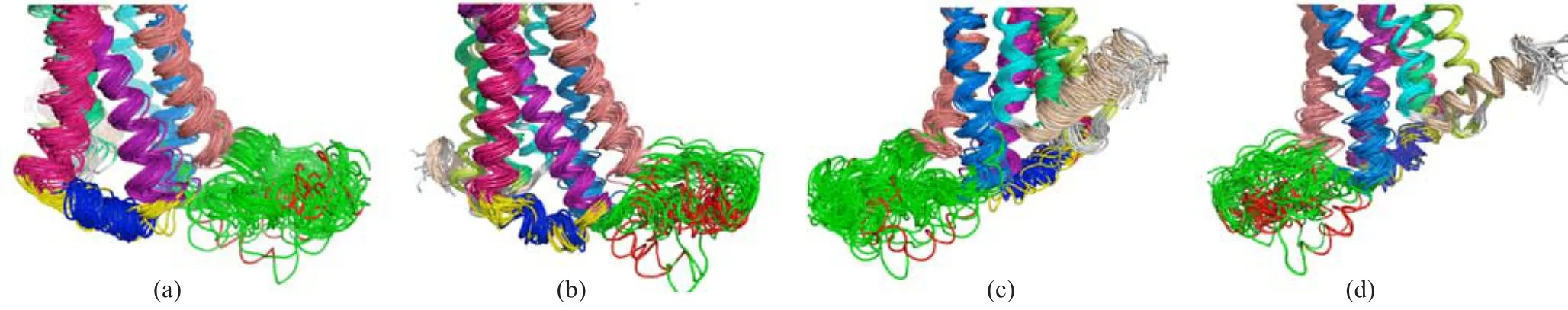
FIG.10 Secondary structures of apo-A2AAR and adenosine-A2AAR shown in cartoon with TMs colored in rainbow(TM1 limon,TM2 green,TM3 purple,TM4 pink,TM5 salmon,TM6 marine,TM7 cyan and TM8 wheat).For IC2,α-helix is colored in blue and irregular loops colored in yellow.For IC3,α-helix is colored in red while irregular loops colored in green. (a)In apo-A2AAR,IC2 formed quite stable α-helix while IC3 is a fl exible loop.(b)In adenosine-A2AAR,IC2 samples fl exible loops more frequently and IC3 formed α-helixes.(c)and(d)The back view of apo-A2AAR and adenosine-A2AAR, respectively.
IV.CONCLUSION
In this work,the adenosine binding to A2AAR and its induced conformational dynamics were studied.Firstly we investigated the agonist binding and identif i ed key interacting residues by binding energy calculation and its decomposition.Besides H2506.52and N2536.55, A632.61,I662.64,V843.32,L853.33,T883.36,F1685.29, M1775.38and L2496.51were also detected to interact with adenosine,which was consistent with the recently published adenosine-A2AAR crystal structure. Moreover,no strong interaction between adenosine and L1675.28was observed,which agreed with our previous fi ndings that L1675.28is an antagonist speci fi c binding reside[36].Thus,enhancing the antagonist binding with L1675.28could possibly trigger the A2AAR inactivation,and agonist interacting with L1675.28could possibly lower the activating efficiency.
Furthermore,wedemonstratedthecharacteristic functional switches:ionic lock,rotamer toggle switch and IC2/IC3,by comparing apo-A2AAR with adenosine-A2AAR.Tight ionic lock between TM3 and TM6 formed in adenosine-A2AAR,while the ionic lock in apo-A2AAR equilibrated between lock and unlock comformations.The rotamer toggle switch, T883.36/F2426.44/W2466.48,adopted different χ1 rotation states in apo-A2AAR and adenosine-A2AAR.Besides,apo-A2AAR adopted α-helical IC2 and f l exible IC3,while adenosine-A2AAR showed f l exible IC2 and α-helical IC3.
Together with our previous work on antagonist bound A2AARs[36],the results suggested that agonist/antagonist bound A2AARs had similar rotamer toggle switch conformation and secondary structures of IC2/IC3,which were different from apo-A2AAR.Meanwhile,tight ionic lock was formed in agonist-A2AAR, but it was broken in antagonist bound A2AAR and equilibrated between formation and breakage in apo-A2AAR.Thus,the agonism/antagonism dynamic behaviors of these switches could be used as monitors of A2AAR activation/inactivation transition and help to unravel the functional mechanisms of A2AAR.
Supplementary material:Ballesteros-Weinstein residue numbering methods are shown as follows.For GPCRs,in addition to numbering the residues by their positions in the primary amino acid sequence, the residues have also been numbered in superscripts (X.YY)that indicate their position in each transmembrane helix(X,helix number,from 1 to 8)relative to the most conserved reference residue in that helix(YY). This residue is arbitrarily assigned the number 50,and numbers decrease toward the N-terminus and increase toward the C-terminus.However,the numbering is not used in loop regions beyond residues X.20 and/or X.80 or T4L.For example,W2466.48is the 246th amino acid in A2AAR,and it is 2 residues N-terminus adjacent to the most conserved reside P2486.50in TM6.
[1]M.A.Hanson and R.C.Stevens,Structure 17,8 (2009).
[2]J.Zezula and M.Freissmuth,Br.J.Pharmacol.153, S184(2008).
[3]J.A.Beavo and L.L.Brunton,Nat.Rev.Mol.Cell Biol.3,710(2002).
[4]V.Ramkumar,D.M.Hallam,and Z.Z.Nie,Jpn.J. Pharmacol.86,265(2001).
[5]J.Linden,Annu.Rev.Pharmacol.41,775(2001).
[6]M.V.Sitkovsky,D.Lukashev,S.Apasov,H.Kojima, M.Koshiba,C.Caldwell,A.Ohta,and M.Thiel,Annu. Rev.Immunol.22,657(2004).
[7]S.Majumdar,and B.B.Aggarwal,Oncogene 22,1206 (2003).
[8]Q.Liu,J.Li,J.Khoury,S.P.Colgan,and J.C.Ibla, J.Biol.Chem.284,13686(2009).
[9]J.Khoury,J.C.Ibla,A.S.Neish,and S.R.Colgan,J. Clin.Invest.117,703(2007).
[10]F.Xu,H.X.Wu,V.Katritch,G.W.Han,K.A.Jacobson,Z.G.Gao,V.Cherezov,and R.C.Stevens, Science 332,322(2011).
[11]G.Lebon,T.Warne,P.C.Edwards,K.Bennett,C. J.Langmead,A.G.W.Leslie,and C.G.Tate,Nature 474,521(2011).
[12]T.Hino,T.Arakawa,H.Iwanari,T.Yurugi-Kobayashi, C.Ikeda-Suno,Y.Nakada-Nakura,O.Kusano-Arai,S. Weyand,T.Shimamura,N.Nomura,A.D.Cameron, T.Kobayashi,T.Hamakubo,S.Iwata,and T.Murata, Nature 482,237(2012).
[13]A.S.Dore,N.Robertson,J.C.Errey,I.Ng,K.Hollenstein,B.Tehan,E.Hurrell,K.Bennett,M.Congreve, F.Magnani,C.G.Tate,M.Weir,and F.H.Marshall, Structure 19,1283(2011).
[14]V.P.Jaakola,M.T.Griffith,M.A.Hanson,V.Cherezov,E.Y.T.Chien,J.R.Lane,A.P.Ijzerman,and R. C.Stevens,Science 322,1211(2008).
[15]J.Y.Lee and E.Lyman,Biophys.J.102,2114(2012).
[16]E.Lyman,C.Higgs,B.Kim,D.Lupyan,J.C.Shelleys, R.Farid,and G.A.Voth,Structure 17,1660(2009).
[17]P.Scheerer,J.H.Park,P.W.Hildebrand,Y.J.Kim,N. Krauss,H.W.Choe,K.P.Hofmann,and O.P.Ernst, Nature 455,497(2008).
[18]D.L.Farrens,C.Altenbach,K.Yang,W.L.Hubbell, and H.G.Khorana,Science 274,768(1996).
[19]S.P.Sheikh,T.A.Zvyaga,O.Lichtarge,T.P.Sakmar, and H.R.Bourne,Nature 383,347(1996).
[20]R.Singh,D.P.Hurst,J.Barnett-Norris,D.L.Lynch, P.H.Reggio,and F.Guarnieri,J.Pept.Res.60,357 (2002).
[21]L.Shi,G.Liapakis,R.Xu,F.Guarnieri,J.A.Ballesteros,and J.A.Javitch,J.Biol.Chem.277,40989 (2002).
[22]K.Palczewski,T.Kumasaka,T.Hori,C.A.Behnke, H.Motoshima,B.A.Fox,I.Le Trong,D.C.Teller, T.Okada,R.E.Stenkamp,M.Yamamoto,and M. Miyano,Science 289,739(2000).
[23]T.Warne,M.J.Serrano-Vega,J.G.Baker,R. Moukhametzianov,P.C.Edwards,R.Henderson,A. G.Leslie,C.G.Tate,and G.F.Schertler,Nature 454, 486(2008).
[24]E.S.Burstein,T.A.Spalding,and M.R.Brann,J. Biol.Chem.273,24322(1998).
[25]S.K.F.Wong,E.M.Parker,and E.M.Ross,J.Biol. Chem.265,6219(1990).
[26]J.Wess,T.I.Bonner,F.Dorje,and M.R.Brann,Mol. Pharmacol.38,517(1990).
[27]S.K.F.Wong,and E.M.Ross,J.Biol.Chem.269, 18968(1994).
[28]J.F.Shan,H.Weinstein,andE.L.Mehler, Biochemistry-US 49,10691(2010).
[29]M.Ambrosio,A.Zurn,and M.J.Lohse,Neuropharmacology 60,45(2011).
[30]C.Altenbach,K.Yang,D.L.Farrens,Z.T. Farahbakhsh,H.G.Khorana,and W.L.Hubbell, Biochemistry-US 35,12470(1996).
[31]K.Arnold,L.Bordoli,J.Kopp,and T.Schwede,Bioinformatics 22,195(2006).
[32]D.Eisenberg,R.Luthy,and J.U.Bowie,Macromolecular Crystallography,Pt.B,277,396(1997).
[33]T.A.D.D.A.Case,T.E.Cheatham,III,C.L.Simmerling,J.Wang,R.E.Duke,R.Luo,M.Crowley, R.C.Walker,W.Zhang,K.M.Merz,B.Wang,S. Hayik,A.Roitberg,G.Seabra,I.Kolossva´ary,K.F. Wong,F.Paesani,J.Vanicek,X.Wu,S.R.Brozell,T. Steinbrecher,H.Gohlke,L.Yang,C.Tan,J.Mongan, V.Hornak,G.Cui,D.H.Mathews,M.G.Seetin,C. Sagui,V.Babin,and P.A.Kollman,AMBER 10,San Francisco:University of California(2008).
[34]J.C.Gordon,J.B.Myers,T.Folta,V.Shoja,L.S. Heath,and A.Onufriev,Nucleic Acids Res.33,W368 (2005).
[35]O.Trott and A.J.Olson,J.Comput.Chem.31,455 (2010).
[36]X.Q.Pang,M.J.Yang,and K.L.Han,Proteins Proteins.81,1399(2013).
[37]O.Vajragupta,P.Boonchoong,G.M.Morris,and A. J.Olson,Bioorg.Med.Chem.Lett.15,3364(2005).
[38]W.Humphrey,A.Dalke,and K.Schulten,J.Mol. Graphics.14,33(1996).
[39]J.C.Phillips,R.Braun,W.Wang,J.Gumbart,E. Tajkhorshid,E.Villa,C.Chipot,R.D.Skeel,L.Kale, and K.Schulten,J.Comput.Chem.26,1781(2005).
[40]F.Y.Dupradeau,C.Cezard,R.Lelong,E.Stanislawiak,J.Pecher,J.C.Delepine,and P.Cieplak,Nucleic Acids Res 36,D360(2008).
[41]G.W.T.M.J.Frisch,H.B.Schlegel,G.E.Scuseria, M.A.Robb,J.R.Cheeseman,J.A.Jr.Montgomery, T.Vreven,K.N.Kudin,J.C.Burant,J.M.Millam, S.S.Iyengar,J.Tomasi,V.Barone,B.Mennucci,M. Cossi,G.Scalmani,N.Rega,G.A.Petersson,H.Nakatsuji,M.Hada,M.Ehara,K.Toyota,R.Fukuda,J. Hasegawa,M.Ishida,T.Nakajima,Y.Honda,O.Kitao, H.Nakai,M.Klene,X.Li,J.E.Knox,H.P.Hratchian, J.B.Cross,V.Bakken,C.Adamo,J.Jaramillo,R. Gomperts,R.E.Stratmann,O.Yazyev,A.J.Austin, R.Cammi,C.Pomelli,J.W.Ochterski,P.Y.Ayala, K.Morokuma,G.A.Voth,P.Salvador,J.J.Dannenberg,V.G.Zakrzewski,S.Dapprich,A.D.Daniels,M. C.Strain,O.Farkas,D.K.Malick,A.D.Rabuck,K. Raghavachari,J.B.Foresman,J.V.Ortiz,Q.Cui,A. G.Baboul,S.Clifford,J.Cioslowski,B.B.Stefanov,G. Liu,A.Liashenko,P.Piskorz,I.Komaromi,R.L.Martin,D.J.Fox,T.Keith,M.A.Al-Laham,C.Y.Peng, A.Nanayakkara,M.Challacombe,P.M.W.Gill,B. Johnson,W.Chen,M.W.Wong,C.Gonzalez,and J. A.Pople,Gaussian 03,Revision C.02,Wallingford CT: Gaussian,Inc.,(2004).
[42]T.Wang and Y.Duan,J.Am.Chem.Soc.129,6970 (2007).
[43]G.J.Martyna,D.J.Tobias,and M.L.Klein,J.Chem. Phys.101,4177(1994).
[44]S.E.Feller,Y.H.Zhang,R.W.Pastor,and B.R. Brooks,J.Chem.Phys.103,4613(1995).
[45]J.P.Ryckaert,G.Ciccotti,and H.J.C.Berendsen,J. Comput.Phys.23,327(1977).
[46]T.Darden,D.York,and L.Pedersen,J.Chem.Phys. 98,10089(1993).
[47]H.Y.M.Chen,X.X.Lin,Y.T.Chen,M.L.Wang, and J.H.Liu,Commun.Comput.Chem.1,72(2013). [48]Y.Q.Jing and K.L.Han,Expert Opin.Drug Discovery 5,33(2010).
[49]Y.W.D.M.Li,and K.L.Han,Coord.Chem.Rev. 256,1137(2012).
[50]M.J.Yang,X.Q.Pang,X.Zhang,and K.L.Han,J. Struct.Biol.173,57(2010).
[51]W.Kabsch and C.Sander,Biopolymers 22,2577 (1983).
[52]J.H.Kim,J.Wess,A.M.Vanrhee,T.Schoneberg,and K.A.Jacobson,J.Biol.Chem.270,13987(1995).
[53]W.H.Ballesteros,Methods Neurosci.25,366(1995).
[54]J.H.Park,P.Scheerer,K.P.Hofmann,H.W.Choe, and O.P.Ernst,Nature 454,183(2008).
[55]S.G.Rasmussen,H.J.Choi,D.M.Rosenbaum,T. S.Kobilka,F.S.Thian,P.C.Edwards,M.Burghammer,V.R.Ratnala,R.Sanishvili,R.F.Fischetti,G.F. Schertler,W.I.Weis,and B.K.Kobilka,Nature 450, 383(2007).
[56]T.Higashijima,S.Uzu,T.Nakajima,and E.M.Ross, J.Biol.Chem.263,6491(1988).
[57]J.Lechleiter,R.Hellmiss,K.Duerson,D.Ennulat,N. David,D.Clapham,and E.Peralta,EMBO J.9,4381 (1990).
ceived on July 29,2013;Accepted on July 22,2013)
∗Author to whom correspondence should be addressed.E-mail:beam@dicp.ac.cn,Tel.:+86-411-84379195,FAX:+86-411-84675584
 CHINESE JOURNAL OF CHEMICAL PHYSICS2014年1期
CHINESE JOURNAL OF CHEMICAL PHYSICS2014年1期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Experimental and Theoretical Investigation on Excited State Intramolecular Proton Transfer Coupled Charge Transfer Reaction of Baicalein
- Theoretical Study of Reagent Rotational Excitation effect on the Stereodynamics of H+LiF→HF+Li Reaction
- Infrared Spectroscopy of COIsolated in Solid Nitrogen Matrix
- effects of Rotational Isomerism and Bond Length Alternation on Optical Spectra of FTC Chromophore in Solution
- High Pressure Structural Instability and Thermal Properties of Rutile TiO2from First-principles
- Kinetic Implication from Temperature effect on Hydrogen Evolution Reaction at Ag Electrode