
Experimental and Theoretical Investigation on Excited State Intramolecular Proton Transfer Coupled Charge Transfer Reaction of Baicalein
2014-07-19 11:17:12ShanshanHuKunLiuQianqianDingWeiPengMaoduChen
Shan-shan Hu,Kun Liu,Qian-qian Ding,Wei Peng,Mao-du Chen
School of Physics and Optoelectronic Technology,and College of Advanced Science and Technology, Dalian University of Technology,Dalian 116024,China
Experimental and Theoretical Investigation on Excited State Intramolecular Proton Transfer Coupled Charge Transfer Reaction of Baicalein
Shan-shan Hu,Kun Liu,Qian-qian Ding,Wei Peng∗,Mao-du Chen∗
School of Physics and Optoelectronic Technology,and College of Advanced Science and Technology, Dalian University of Technology,Dalian 116024,China
The excited state intramolecular proton transfer(ESIPT)coupled charge transfer of baicalein has been investigated using steady-state spectroscopic experiment and quantum chemistry calculations.The absence of the absorption peak from S1excited state both in the experimental and calculated absorption spectra indicates that S1is a dark state.The dark excited state S1results in the very weak fluorescence of solid baicalein in the experiment.The frontier molecular orbital and the charge difference densities of baicalein show clearly that the S1state is a charge-transfer state whereas the S2state is a locally excited state.The only one stationary point on the potential energy profile of excited state suggests that the ESIPT reaction of baicalein is a barrierless process.
Excited state intramolecular proton transfer,Intramolecular charge transfer, Time-dependent density functional theory,Dark state,Baicalein
I.INTRODUCTION
Proton transfer is a process in which a proton transfers from one atom to another atom in the ground or the excited state.This process can be depicted as a structure transformation from A-H···B to A···H-B, which takes place via a hydrogen bond[1].The excited state intramolecular proton transfer(ESIPT)occurs in the electronic excited state of a molecule for which a heterocyclic ring is formed by the intramolecular hydrogen bond between a hydroxyl group and a neighboring proton acceptor[2].ESIPT has gained considerable attentions from both theoretical and experimental perspectives due to its extensive application in chemical and biochemical f i eld,ever since the 1950s when Albert Weller laid foundation for it[2-18].The molecule which undergoes the ESIPT process exhibits characteristic photophysical features such as ultrafast reaction time which are commonly attributed to a barrierless process,or at least one with a very low barrier,the 4-level reaction scheme,and large Stokes shift[2].Because of these features,it can be exploited in luminescent materials,proton transfer laser[3,4],molecular logic gate[5],UV stabilizers[6],and solar concentrators[7]among others.The extreme sensitivity makes it possible to be a promising application f i eld in fluorescence sensors and probes for macromolecular science and cellular biology[8-10].Moreover,it has also wide applications in lasing dyes[11],photostabilizers[12], Raman filters,and hard-scintillation counters[13],energy/date storage and optical switching[14,15],and triplet quenchers[16].With the deepening of the research,the charge transfer is often found in ESIPT and has been paid more concerns.
The proton coupled charge transfer in the excited state plays a crucial role in a wide range of chemical and biological processes,so it has been widely studied by experiments and theoretical calculations[3,19-21]. Kiefer et al.have found that the intermolecular intrinsic barrier is largely determined by solvent reorganization because a charge redistribution is involved[19]. The coupling of proton motion with charge separation is crucial for most of the important processes in biological energy conversion,such as transmission of nervous impulses,respiration and photosynthesis[21].The application of the fast and efficient fluorescence probe, such as laurdan in the biomolecular fluorescence imaging,also utilizes intramolecular charge transfer(ICT) [22].In addition,Zhao and Han have explained the drastic fluorescence quenching behaviors of oxazine 750 (OX750)chromophore in protic alcoholic by a solutionsolvent intermolecular photoinduced electron transfer reaction[23].Recently,Chou and his colleagues made a detailed review of the ESPT coupled CT reaction, which could be modulated by molecular structure and media polarization[24].
The ESIPT reaction has a wide range of applications,but the applications in medicine are rarely found, because of the tendency to focus on the medicines’medicinal properties while ignoring their photochemical reaction,despite their photochemical and photophysical properties contribution to the medicinal prop-erties.Baicalein is a typical fl avonoid compound from the root of Scutelluriu buicalensis,which is a kind of traditional Chinese herbal medicine.It is known to have anti-allergic,anti-carcinogenic,anti-HIV,antioxidative,anti-in fl ammatory,anti-viral properties,and can be used to treat hepatitis,bronchitis,nephritis, asthma,atopic dermatitis,cirrhosis,jaundice,hepatoma,leukemia,hyperlipemia,arteriosclerosis,and infl ammatory diseases[25-29].Consequently,its pharmacological e ff ects have been a topic of interest for years.However,its photophysical and photochemical properties have not been studied deeply so far.It is known that the intrinsically fl uorescent properties (such as doxorubicin)of drugs make them convenient for probing and visualization with various microscopic imaging technologies[30],which will greatly contribute to the drugs action mechanism and pharmacokinetics. Therefore,the current study of photophysical and photochemical properties of medicines will also provide a promising pathway to the development of new drugs based on traditional Chinese medicine monomers.
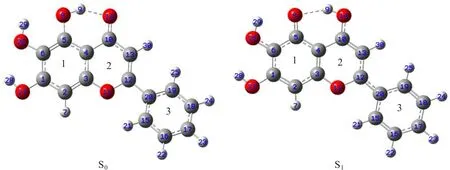
FIG.1 The geometric structures of baicalein in the ground state(S0normal form)and excited state(S1tautomer form),1, 2,and 3 stand for the f i rst,second and third benzene ring respectively.
In this work,we mainly investigate the excited state intramolecular proton transfer coupled charge transfer of baicalein using the DFT/TDDFT and experiment methods.The bond lengths and bond angles of baicalein in both the ground and excited states are analyzed and their potential energy pro fi les are discussed. The calculated and experimental absorption and fl uorescence spectra are displayed and discussed.The charge transfer of baicalein is demonstrated by analyzing the frontier molecular orbits and the charge di ff erence densities(CDDs).The elaborated study of spectra and photoinduced proton and charge transfer process gives insight into the photophysical and photochemical properties of traditional Chinese herbal medicine.
II.EXPERIMENTAL AND COMPUTATIONAL DETAILS
For the experiment,the absorption spectrum of baicalein in ethanol solution(10µmol/L)is measured by the MAYA2000PRO Spectrometers.The fluorescence spectrum of solid baicalein is obtained using the RF5301PC Spectrometers(SHIMADZU Corp.)at excitation wavelength of 325 nm.It is worth noting that all the experiments are performed at room temperature.
The geometric structure and properties of baicalein in the ground state are recorded using the DFT method. The geometric structure and properties of excited state are calculated by means of the TDDFT method.The TDDFT method has been conf i rmed as an useful tool to investigate the properties of the excited state by many theoretical investigations[17,18,31-35].The Becke’s three-parameter hybrid exchange function with Lee-Yang-Parr gradient-corrected correlation functional (B3LYP)[36]and the triple-ζ valence quality with one set of polarization functions(TZVP)[37]are chosen as functional and basis sets throughout calculations. Fine quadrature grids 4[38]are also employed.The default self-consistent f i eld(SCF)convergency threshold of the energy(10-6)is used for the optimization of the ground and excited states geometries.All electronic structure calculations of baicalein are completed without constraints for symmetry.In addition,it is confi rmed that all of the local minima are in the absence of an imaginary mode through the calculation of vibrational frequency.In order to reproduce the experimental results,the solvent e ff ect in ethanol solution is employed in the absorption spectrum calculation by using the conductor-like screening model(COSMO)method. The calculation of fl uorescence spectrum of baicalein is conducted under the condition of vacuum environment. All the calculations in this work are preformed with the TURBOMOLE program suite[36-38].The transition densities(TDs)and the charge di ff erence densities (CDDs)methods are used to describe the characteristics of transition dipole moments and charge transfer [39-41].
III.RESULTS AND DISCUSSION
The geometric structures of baicalein in the ground and excited states are optimized to depict the in-tramolecular proton transfer reaction of baicalein,and the results are shown in Fig.1.It is revealed that the proton transfers from O-H to C=O upon the excitation of the ground state to its excited state.That is, in the ground state,the enol isomer of baicalein(normal form)is more stable than the keto isomer(tautomer form),while the relative stability is reversed in the excited state.This demonstrates that the baicalein undergoes an ultrafast ESIPT process through the sixmembered ring which consists of the intramolecular hydrogen bond O-H···O=C once being excited.It is worthwhile to mention that the stable geometric structure of the ground state is non-planar and the dihedral angle of DO11C12C20C15is-18.05◦,but it becomes planar structure with a very small dihedral angle of 0.05◦after being excited.This suggests that upon the excitation of the normal form to its f i rst-excited singlet state,the enhanced acidity of the hydroxyl group causes an ultrafast ESIPT from this group to the neighboring carbon-based,affording the planar tautomer.
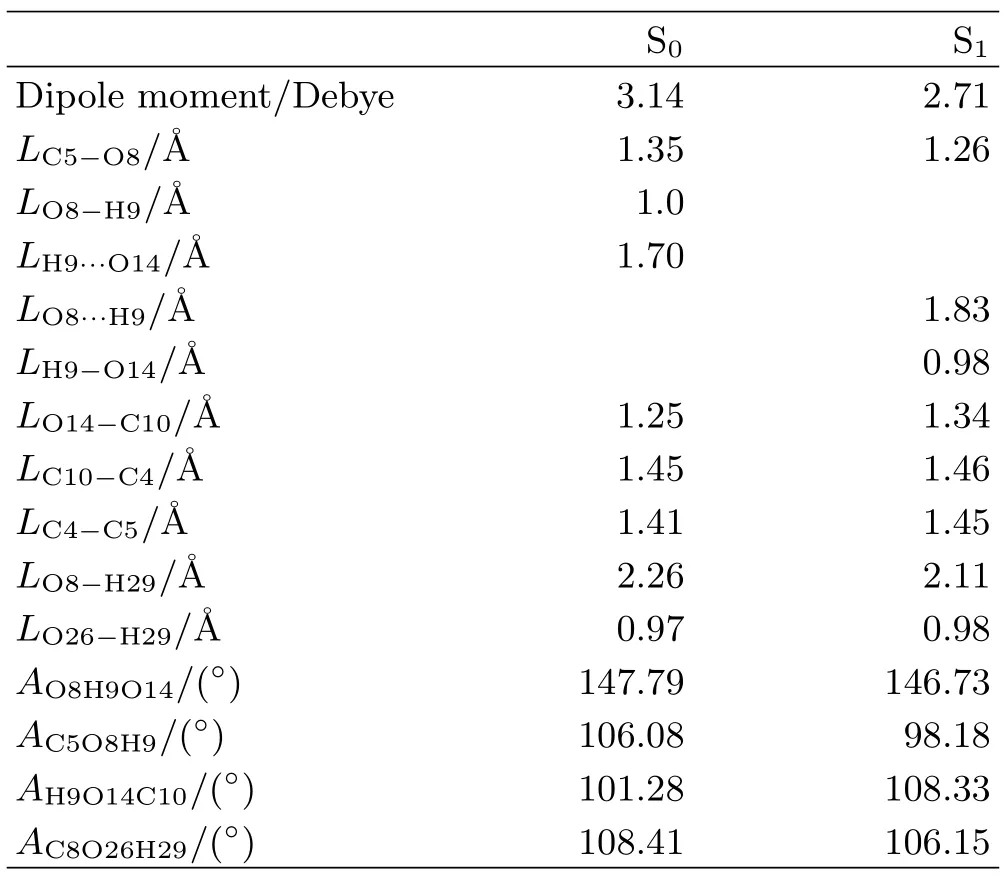
TABLEI The bond lengths L,bond angles A,and dipole moments of baicalein in the ground state and excited state.

FIG.2(a)The experimental absorption spectrum in ethanol solution and the calculated absorption spectrum with the consideration of the COSMO solvation model.(b) The experimental fluorescence spectrum of solid baicalein and the calculated fluorescence spectrum in the vacuum environment.
To further depict the ESIPT reaction of baicalein, we list the changes of the bond lengths and bond angles of baicalein in the ground and excited states in Table I.From the ground state to the excited state,the bond length LO8-H9increases from 1.0˚A to 1.83˚A and LH9-O14decreases from 1.7˚A to 0.98˚A. The bond angle AO8H9O14decreases from 147.79◦to 146.73◦.The changes of bond lengths and bond angle manifest that the intramolecular hydrogen bond O8-H9···O14 breaks,from the ground state to the excited state,accompanying with the formation of an intramolecular hydrogen bond O8···H9-O14.The bond lengths LC5-O8,LC10-O14,LC4-C5,LC10-C4as well as the bond angles AH9O8C5and AH9O14C10also change because of the influence of the intramolecular proton transfer.The intramolecular hydrogen bond O8-H9···O14 in the ground state is 1.70˚A,but in the excited state the intramolecular hydrogen bond O8···H9-O14 increases to 1.83˚A.The increase of the length of intramolecular hydrogen bond reveals that the intramolecular hydrogen bond in the excited state is weaker than that in the ground state,because O8 is affected by H29 when H9 transfers to O14 in the excited state.The bond length LO8-H29,LO26-H29,and bond angle AC8H29O26are also listed in Table I.From the ground state to the excited state,the LO26-H29increases by 0.01˚A,but the LO8-H29decreases by 0.15˚A.At the same time,the AC8H29O26decreases by 2.26◦.These changes show that the O8 moves to H29 upon the excitation of baicalein to its excited state.Thus,it makes the intramolecular hydrogen bond O8···H9-O14 longer in the excited state.In addition, the data of the dipole moment in the ground and excited states are summarized in Table I.In the excited state,it is 2.71 Debye which is smaller than that in the ground state.This demonstrates that the variation of electron density on the ground state is more obvious than that in the excited state.
Figure 2 displays the experimental and calculated ab-sorption and fluorescence spectra of baicalein.It is worth noting that the experimental absorption spectrum is measured in the ethanol solution and the calculated absorption spectrum is recorded with considering the solvent effects in ethanol.From Fig.2(a)we can see that the calculated absorption peaks are located at about 323 and 276 nm,which agree well with the experiment values 325 and 275 nm,respectively.It can also be seen that the absorption peak of 325 nm in the experiment corresponds to the calculated result 323 nm, which comes from the S2state according to our calculation.At the same time,we also f i nd that the calculated S1state has a small oscillator strength 0.029, which makes it clear that S1is a dark state.As a result,the absorption peak of the S1state is not observed experimentally.Thus,it can be introduced that when the baicalein is excited,the electrons in the ground state will have a transition to the S2state,and then go through the internal conversion process to reach the S1state,but at last revert back to the S0state mainly through some nonradiative transition process which will result in the weak fluorescence of baicalein.For comparison,we also measure the fluorescence of baicalein in ethanol solution,but it is absent.Figure 2(b)is the experimental fluorescence spectrum of solid baicalein and the calculated fluorescence spectrum in the vacuum environment.It clearly shows that the fluorescence peak of solid baicalein is very weak.This is coincident with the above discussion.Therefore,both the experiment and theoretical calculation indicate that S1state is a dark state.It can be also found that the calculated fl uorescence peak of 446 nm is in accordance with the experimental value of 468 nm.
It is known to all that the frontier molecular orbitals analysis can provide insight into the nature of excited state[18,42-44].According to our calculation,it is known that the S1state is mainly the transition from HOMO to LUMO and the S2state is mainly the transition from HOMO-1 to LUMO.Thus,only the HOMO-1, HOMO,and LUMO orbitals of baicalein in the ground state(normal form)and the excited state(tautomer form)are displayed in Fig.3.We can fi nd that the electron density of HOMO is mainly localized on the fi rst benzene ring either in the ground or in the excited state of baicalein,but the electron densities of both the HOMO-1 and LUMO spread all over the molecule.So, it is con fi rmed that S1state is of ICT character.The charge can be transferred from the fi rst benzene ring to the second and third benzene ring.However,S2state is a locally excited state because the electron densities of both the HOMO-1 and LUMO spread across the molecule.From Fig.3,it is evident that the S1and S2states are both of ππ∗character.In addition, from the frontier molecular orbitals of normal form, when we only focus on the electron density of hydroxide group and O14,it can be found that the electron density of HOMO mainly locates on the hydroxide group while HOMO-1 has an uniform distribution on hydroxide group and O14.For LUMO,it mainly locates on the O14.Therefore,upon the excitation of the normal form to its excited state,the ESIPT occurs more easily in the S1state.

FIG.3 The frontier molecular orbitals of baicalein in the ground state(normal form)and excited state(tautomer form).

FIG.4 TDs and CDDs of baicalein in the S1and S2states. The green and red colors stand for the hole and the electron, respectively,and the isovalue is 1×10-4a.u.
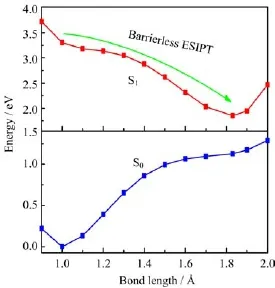
FIG.5 The potential-energy profiles of baicalein along the bond O8-H9 in the ground state S0and excited state S1.
Figure 4 is the TDs and CDDs of baicalein in the S1and S2states.From the TDs of the S1state,we can clearly see that the distribution of electrons and holes are disorganized and then transition dipole moments of each unit cancel each other,which results in a small transition dipole moment of the S1state.This is the reason that the absorption peak of the S1state can’t be observed in both the experiment and calculation.For the S2state,the holes mainly distribute in the six-membered ring which consists of the intramolecular hydrogen bond O-H···O=C,but the electrons mainly distribute in the f i rst benzene ring.The well-organized distribution of electrons and holes illustrates that the transition of the S2state is strong,so the absorption peak of the S2state can be observed.The analysis of the TDs of the S1and S2states also indicates that S1is a dark state.In order to clarify the distribution of the holes and the electrons in the S1and S2states, the CDDs of them are also given.The CDDs of the S1state show that the holes mainly distribute in the fi rst benzene ring,but the electrons are mainly in the second and third benzene ring.It means that the electrons will transfer from the fi rst benzene ring to the second and third benzene rings when the baicalein is excited to its S1state.This also demonstrates that the dark state of the S1is a charge-transfer state.Likewise, it is known that S2is a locally excited state because the CDDs of the S2state show that both the electrons and holes distribute in every benzene ring.The above calculations indicate that the reaction of baicalein in the excited state is a process of proton transfer coupled charge transfer.
To further investigate the properties in the excited state of baicalein,the potential energy pro fi les of baicalein in both the ground and excited states along with the bond O8-H9 are studied,which are shown in Fig.5.It shows that there is only one stationary point either in the ground state or the excited state and we can fi nd that the most stable geometrics in the gr˚oundstate and excited state are at about 0.99 and 1.82A,respectively.Therefore,it can be concluded that only the enol isomer exhibits the stable geometric structure in the ground state but in the excited state only the keto isomer is stable.By observing the excited state potential energy pro fi les of baicalein along with the bond O8-H9,it can be concluded that its excited state has no energy barriers.Therefore,the process of ESIPT reaction is barrierless.
IV.CONCLUSION
In this work,the DFT/TDDFT methods combined with the experiment method are used to investigate the ESIPT coupled charge transfer reaction of baicalein. The optimized geometric structures of baicalein suggest that it undergoes an ESIPT process through the six-membered ring which consists of the intramolecular hydrogen bond O-H···O=C upon the excitation of the ground state to its excited state.The analysis of bond lengths and bond angles of baicalein demonstrates that the intramolecular hydrogen bond in the excited state is weakened.The calculated absorption spectrum indicates that S1is a dark state and is absent in the experiment.Experimental observation of absorption peak 325 nm is the S2state,and the TDs conf i rm that it is a local excited state.Furthermore,TDs and CDDs also declare that the S1is a dark state with ICT character.The fluorescence of solid baicalein is very weak because of the dark S1state.The potential energy profiles of baicalein illustrate that the ESIPT is a barrierless process and only keto form is stable in the excited state.
V.ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.61137005 and No.10974023),the Program for Liaoning Excellent Talents in University(No.LJQ2012002),and the Program for New Century Excellent Talents in University (No.NCET-12-0077).
[1]K.Das,N.Sarkar,A.K.Ghosh,D.Majumdar,D.N. Nath,and K.Bhattacharyya,J.Phys.Chem.98,9126 (1994).
[2]C.H.Kim,J.Park,J.Seo,S.Y.Park,and T.Joo,J. Phys.Chem.A 114,5618(2010).
[3]C.C.Hsieh,C.M.Jiang,and P.T.Chou,Acc.Chem. Res.43,1364(2010).
[4]J.E.Kwon and S.Y.Park,Adv.Mater.23,3615 (2011).
[5]V.Luxami and S.Kumar,New J.Chem.32,2074 (2008).
[6]J.Keck,H.E.A.Kramer,H.Port,T.Hirsch,P.Fischer,and G.Rytz,J.Phys.Chem.100,14468(1996).
[7]D.Y.Chen,C.L.Chen,Y.M.Cheng,C.H.Lai,J.Y. Yu,B.S.Chen,C.C.Hsieh,H.C.Chen,L.Y.Chen,C. Y.Wei,C.C.Wu,and P.T.Chou,ACS Appl.Mater. Interfaces 2,1621(2010).
[8]S.Y.Park,H.Jeong,H.Yu,S.Y.Park,and D.J.Jang, Photochem.Photobiol.86,1197(2010).
[9]K.Y.Chen,Y.M.Cheng,C.H.Lai,C.C.Hsu,M. L.Ho,G.H.Lee,and P.T.Chou,J.Am.Chem.Soc. 129,4534(2007).
[10]A.S.Klymchenko and A.P.Demchenko,J.Am.Chem. Soc.124,12372(2002).
[11]P.T.Chou,M.L.Martinez,and J.H.Clements,J. Phys.Chem.97,2618(1993).
[12]D.B.O’Connor,G.W.Scott,D.R.Coulter,and A. Yavrouian,J.Phys.Chem.95,10252(1991).
[13]M.L.Martinez,W.C.Cooper,and P.T.Chou,Chem. Phys.Lett.193,151(1992).
[14]K.Kuldov´a,A.Corval,H.P.Trommsdorf f,and J.M. Lehn,J.Phys.Chem.A 101,6850(1997).
[15]P.T.Chou,D.McMorrow,T.J.Aartsma,and M. Kasha,J.Phys.Chem.88,4596(1984).
[16]R.M.Tarkka,X.Zhang,and S.A.Jenekhe,J.Am. Chem.Soc.118,9438(1996).
[17]X.H.Zhao and M.D.Chen,Chem.Phys.Lett.512, 35(2011).
[18]X.H.Zhao,Y.F.Liu,L.F.Zhou,Y.Z.Li,and M.D. Chen,J.Lumin.130,1431(2010).
[19]P.M.Kiefer and J.T.Hynes,J.Phys.Chem.A 106, 1850(2002).
[20]X.H.Zhao and M.D.Chen,J.Phys.Chem.A 114, 7786(2010).
[21]S.R´ıos Vzquez,M.C.R´ıos Rodr´ıguez,M.Mosquera, and F.Rodr´ıguez-Prieto,J.Phys.Chem.A 111,1814 (2007).
[22]T.Parasassi,E.Krasnowska,L.Bagatolli,and E.Gratton,J.Fluoresc.8,365(1998).
[23]G.J.Zhao,J.Y.Liu,L.C.Zhou,and K.L.Han,J. Phys.Chem.B 111,8940(2007).
[24]A.P.Demchenko,K.C.Tang,and P.T.Chou,Chem. Soc.Rev.42,1379(2013).
[25]M.Zhu,S.Rajamani,J.Kaylor,S.Han,F.Zhou,and A.L.Fink,J.Biol.Chem.279,26846(2004).
[26]M.Y.Lai,S.L.Hsiu,S.Y.Tsai,Y.C.Hou,and P.D. L.Chao,J.Pharm.Pharmacol.55,205(2003).
[27]B.Q.Li,T.Fu,W.H.Gong,N.Dunlop,H.F.Kung,Y. Yan,J.Kang,and J.M.Wang,Immunopharmacology 49,295(2000).
[28]M.L.Weber,Cancer Treat.Rev.35,57(2009).
[29]Y.C.Chen,S.C.Shen,L.G.Chen,T.J.Lee,and L. L.Yang,Biochem.Pharmacol.61,1417(2001).
[30]D.K.Rana,S.Dhar,A.Sarkar,and S.C.Bhattacharya,J.Phys.Chem.A 115,9169(2011).
[31]G.J.Zhao and K.L.Han,J.Phys.Chem.A 113,4788 (2009).
[32]M.T.Sun,J.Chem.Phys.124,054903(2006).
[33]G.J.Zhao and K.L.Han,J.Phys.Chem.A 111,2469 (2007).
[34]G.J.Zhao and K.L.Han,J.Phys.Chem.A 113,14329 (2009).
[35]G.J.Zhao and K.L.Han,J.Comput.Chem.29,2010 (2008).
[36]A.D.Becke,J.Chem.Phys.98,5648(1993).
[37]A.Schafer,C.Huber,and R.Ahlrichs,J.Chem.Phys. 100,5829(1994).
[38]O.Treutler and R.Ahlrichs,J.Chem.Phys.102,346 (1995).
[39]M.T.Sun,L.W.Liu,Y.Ding,and H.X.Xu,J.Chem. Phys.127,084706(2007).
[40]M.T.Sun,P.Kjellberg,W.J.D.Beenken,and T. Pullerits,Chem.Phys.327,474(2006).
[41]Y.Z.Li,H.X.Li,X.M.Zhao,and M.D.Chen,J. Phys.Chem.A 114,6972(2010).
[42]G.J.Zhao and K.L.Han,Biophys.J.94,38(2008).
[43]G.J.Zhao,B.H.Northrop,K.L.Han,and P.J.Stang, J.Phys.Chem.A 114,9007(2010).
[44]G.J.Zhao and K.L.Han,Acc.Chem.Res.45,404 (2012).
ceived on July 1,2013;Accepted on September 16,2013)
∗Authors to whom correspondence should be addressed.E-mail:wpeng@dlut.edu.cn,mdchen@dlut.edu.cn
 CHINESE JOURNAL OF CHEMICAL PHYSICS2014年1期
CHINESE JOURNAL OF CHEMICAL PHYSICS2014年1期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Theoretical Study of Reagent Rotational Excitation effect on the Stereodynamics of H+LiF→HF+Li Reaction
- Infrared Spectroscopy of COIsolated in Solid Nitrogen Matrix
- effects of Rotational Isomerism and Bond Length Alternation on Optical Spectra of FTC Chromophore in Solution
- High Pressure Structural Instability and Thermal Properties of Rutile TiO2from First-principles
- Kinetic Implication from Temperature effect on Hydrogen Evolution Reaction at Ag Electrode
- Anisotropy of Thermal-expansion for β-Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine:Quantum Chemistry Calculation and Molecular Dynamics Simulation