
Kinetic Implication from Temperature effect on Hydrogen Evolution Reaction at Ag Electrode
2014-07-19 11:17:08JingKangChuhongLinYaoYaoYanxiaChen
Jing Kang,Chu-hong Lin,Yao Yao,Yan-xia Chen
Hefei National Laboratory for Physical Sciences at Microscale,Department of Chemical Physics,University of Science and Technology of China,Hefei 230026,China
Kinetic Implication from Temperature effect on Hydrogen Evolution Reaction at Ag Electrode
Jing Kang,Chu-hong Lin,Yao Yao,Yan-xia Chen∗
Hefei National Laboratory for Physical Sciences at Microscale,Department of Chemical Physics,University of Science and Technology of China,Hefei 230026,China
Hydrogen evolution reaction(HER)at polycrystalline silver electrode in 0.1 mol/L HClO4solution is investigated by cyclic voltammetry in the temperature range of 278-333 K.We found that at electrode potential ϕ<PZC(potential of zero charge),the apparent activation energy Ea,appdecreases with ϕ,while pre-exponential factor A remains nearly unchanged, which conforms well the prediction from Butler-Volmer equation.In contrast,with ϕ negative shifts from the onset potential for HER to the potential of zero charge(PZC≈-0.4 V), both Ea,appand A for HER increase(e.g.,Ea,appincreases from 24 kJ/mol to 32 kJ/mol). The increase in Ea,appand A with negative shift in ϕ from-0.25 V to PZC is explained by the increases of both internal energy change and entropy change from reactants to the transition states,which is correlated with the change in the hydrogen bond network during HER.The positive entropy effects overcompensate the adverse effect from the increase in the activation energy,which leads to a net increase in HER current with the activation energy negative shift from the onset potential of HER to PZC.It is pointed out that entropy change may contribute greatly to the kinetics for electrode reaction which involves the transfer of electron and proton,such as HER.
Hydrogen evolution reaction,Ag electrode,Temperature effect,Activation energy,Pre-exponential factor,Internal energy,Entropy change
I.INTRODUCTION
Hydrogen evolution reaction(HER)is a typical model reaction for understanding the principle of electrocatalysis due to the simplicity of its reaction mechanism[1]. The overall reaction for HER in acidic electrolyte is:
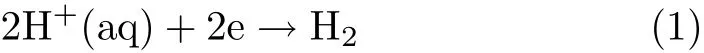
where H+(aq)denotes the hydrated proton,which may be in the form such as H9O4+,H5O2+,or H3O+[2] Depending on the nature of the electrocatalysts,HER can follow two different paths,the Volmer-Tafel or the Volmer-Heyrovsky mechanisms[3],and in both pathways the f i rst step is believed to be the Volmer reaction, i.e.,the discharge of H+,

It may be followed by Heyrovsky reaction,i.e.,an electrochemical desorption step,

Or through the Tafel reaction,i.e.,the combination of two adsorbed H atoms and the subsequent desorption of H2,

The effect of catalyst structure on the HER activity has been extensively studied in the past years,a volcano plot between the rate for HER and the M-H binding energy(EM-H)is well accepted[4,5].From systematic theoretical studies,Norskov et al.suggested that the volcano plot for HER may be better expressed as the rate for HER versus the Gibbs free energy for the H adsorption(GH)at different metal substrates[6], and the best catalysts for HER keep the Gibbs free energy change of all the elementary steps in HER close to zero.Calculations on the activation energy(∆6=Gact) for the rate determining step(rds)for HER at various metal electrodes have also been carried out[7,13-15]. A surprising feature is that the calculated∆6=Gactis remarkably higher than the experimentally measured activation energy,e.g.,from DFT calculation.Norskov et al.found that at Pt(111),∆6=Gactfor HER was ca. 0.85 eV[8],which was much higher than the apparent activation energy(Ea,app)of ca.0.2 eV deduced from temperature-dependent exchange current density reported by Markovic et al.[9].Chen et al.found∆6=Gactfor HER was ca.0.55 eV at Pt(111),whichwas in the middle of the value reported by Norskov and Markovic[10].Schmickler et al.reported that at Cu(111),Ag(111),and Au(111)electrodes,∆6=Gactwas about 0.75 eV[11],while the experimentally measured Ea,appfor HER at Ag(111)from the same group was just in the range from 0.05 eV to 0.25 eV[12].From our recent measurement of temperature effect on HER at quasi-Au(111)electrode,the apparent activation energy is also estimated to be ca.0.2 eV[13].
In theoretical studies,the pre-expoential factor A is very difficult to be calculated,so usually only the activation barrier is used to discuss the activity toward HER [2,11,14-16].Considering the fact that the reactants such as proton and water are located in the hydrogenbonded network,whose structure and configuration are under dynamic changes during HER[17-19],we expect that the entropy change(pre-exponential factor in the Arrhenius equation)may also play a very important role in HER rate.In order to figure out whether such effects are important for HER or not,we have carried out systematic studies of the temperature effect on HER at Pt(111)[20],Ag,Cd,and GC electrodes[21],the parameters,such as apparent activation energy Ea,appand A as well as their potential dependence have been derived.In this work,we report the related data for HER at Ag electrode.
II.EXPERIMENTS
A polycrystalline Ag wire(99.99%,diameter:1 mm, denoted as pc-Ag here after)embedded in a glass holder is used as working electrode(WE).It is polished using Al2O3powder with size of 3,1,and 0.05µm,successively,and before switching to the polishing powder with lower diameter,it is flushed carefully with tape water.After that,it is dipped into 0.1 mol/L HNO3for ca. 10 s,and flushed carefully with Milli-Q water(18.2 MΩ, Milli-Q pure water system).Then,the electrode surface is covered by a water droplet and transferred to the electrochemical cell immediately.A conventional two compartment,three electrodes cell is used in the present study,which is similar to the conventional cell used for RDE system[22],except for an additional glass jacket that allows water circulating around the cell for controlling the cell temperature.An Ag|AgCl(with saturated KCl solution)electrode and an Ag wire are used as reference(RE)and counter electrode(CE),respectively. The potential of the Ag|AgCl(with saturated KCl solution)is-0.256 V vs.reversible hydrogen electrode (RHE)at 298 K.The RE is placed in the second compartment to avoid the contamination of the solution by the leakage of Cl-ions or by traces of dissolved Ag+and it is kept at room temperature(298 K).All potentials in the work are quoted against RHE at 298 K.
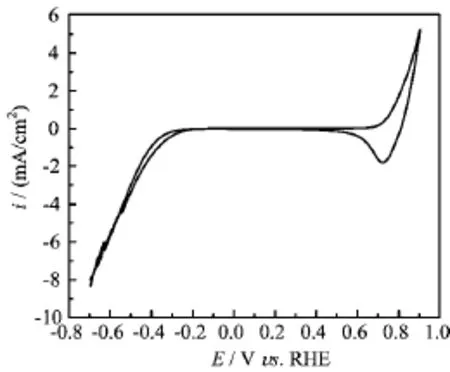
FIG.1 Cyclic voltammogram of Ag electrode in N2saturated 0.1 mol/L HClO4,potential scan rate of 50 mV/s.
The electrolyte solution is 0.1 mol/L HClO4,which is prepared using perchloric acid(70%,Sigma-Aldrich) and ultra-pure water.Before studying the temperature effect,electrode potential is cycled continuously in the potential region from-0.7 V to 0.9 V at a scan rate of 50 mV/s until clean and reproducible cyclic voltammograms(CVs)are obtained.Then,the electrode is washed carefully by ultra-pure water and immediately inserted into the cell filled with newly prepared solutions for HER measurements.When recording the i-E curves for HER,the electrode is rotated at a speed of 500 r/min in order to avoid the interference of H2bubbles on HER kinetics.The rotation of electrode is controlled by a modulated rotator(Hokuto Denko Ltd.). IR compensation has been carried out automatically by the CHI instrument based on positive feed-back principle.The uncompensated Ohmic resistance is measured by AC impedance.During all the measurements,the cell and the atmosphere above the cell are continuously purged with N2(99.999%,Nanjing Special gas,Corp.).
III.RESULTS AND DISCUSSION
A.Cyclic voltammetric characterization of Ag electrode and HER kinetics
Figure 1 displays the cyclic voltammorgams(CV)of Ag electrode in 0.1 mol/L HClO4recorded at room temperature,the pair of redox peaks at ca.0.7 V is due to Ag++e›Ag,and the potential region from-0.1 V to 0.4 V is double layer charging potential regime.At potential negative of-0.15 V,HER occurs,its current density increases monotonically with negative shift in electrode potential.Such features of the CV are quite similar to that reported for Ag/acidic electrolyte system in Ref.[12].The electrochemical active surface area of Ag electrode is estimated to be ca.0.015 cm2(with a roughness factor of ca.2)from the ratio of its double layer capacitance to that of Ag(111),the latter is 40µF/cm2according to Ref.[23].In order to avoid the change of surface roughness of Ag electrode by cycling to higher potentials especially at elevated temperatures, we have only recorded CV up to 0.9 V at room temperature before the measurements for the i-E curves forHER at other temperatures.For the subsequent HER measurements by cyclic voltammetry,the upper potential limit is kept at 0.05 V.
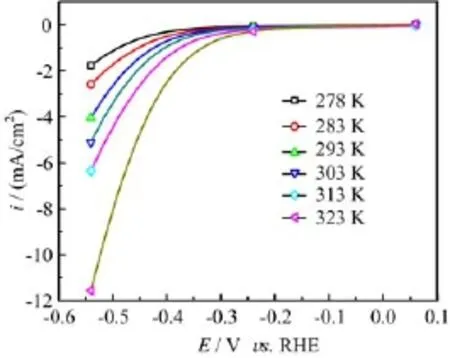
FIG.2 Polarization curves for HER at Ag electrode in N2saturated 0.1 mol/L HClO4at various constant temperatures,potential scan rate of 50 mV/s.
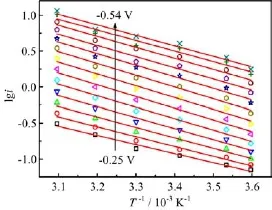
FIG.3 Arrhenius plots for hydrogen evolution reactions at Ag electrode in 0.1 mol/L HClO4at various constant potentials,raw data from Fig.2.
Figure 2 displays the polarization curves for HER in 0.1 mol/L HClO4at various temperatures from 278 K to 323 K.From Fig.2 it is seen that at the same temperature,the HER current increases exponentially toward negative potentials.At f i xed potential the currents display an obvious increase with temperature(the onset potential for HER also increases slightly e.g.,it shifts from-0.2 V at 278 K to ca.-0.15 V at 323 K).All these facts conf i rm that HER at Ag electrode is a fast process.Qualitatively,the temperature and potential dependent HER behavior at Ag electrode is very similar to our previous results on HER at Au electrode [13],except that the onset potential for HER at Ag is ca.0.1 V more negative than that at Au electrode in otherwise identical conditions.

FIG.4 The plots of(a)apparent activation energies(Ea,app) and(b)lnA(pre-expotential factor)for hydrogen evolution reactions at Ag electrode in 0.1 mol/L HClO4as a function of reaction potential,data derived from curves given in Fig.2.
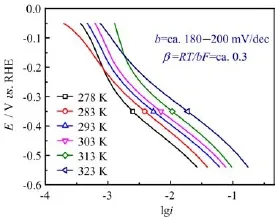
FIG.5 Tafel plots for hydrogen evolution reactions at Ag electrode in N2saturated 0.1 mol/L HClO4at various constant temperatures,data derived from curves given in Fig.2.
The Arrhenius plots for hydrogen evolution reactions at Ag electrode in 0.1 mol/L HClO4at various constant potentials are given in Fig.3,which displays roughly linear behavior and the lines upshift toward more negative potentials.Ea,appand A for HER on pc-Ag at different constant potentials are derived from the slope and the intercept at Y axis of the lines,which are plotted as a function of electrode potential in Fig.4.From Fig.4 it is seen that Ea,appincreases from 24 kJ/mol to 31.5 kJ/mol with electrode potential negative scan from-0.25 V to-0.4 V.And with further negative shift in electrode potential from-0.4 V to-0.55 V, Ea,appdecreases gradually to 28 kJ/mol.On the other hand,with negative potential shift from-0.25 V to -0.40 V,A also displays a monotonically increase,and it remains nearly unchanged at potentials from-0.4 V to-0.55 V.
The Tafel plots for HER at Ag electrode in 0.1 mol/L HClO4at constant temperatures are displayed in Fig.5, from which the Tafel slopes b are found to be ca.180-200 mV/dec in the potential region from-0.2 V to -0.55 V.From the Tafel slope,the transfer coefficient β is found to be ca.0.3 according to Butler-Volmer equation β=RT/bF.Such numbers are very close to those found at Ag(111)[12],which is ca.0.34 to 0.4.It is found that the potential,where the maximum activation energy appears,is just close to the potential of zero charge(PZC)for pc-Ag[23].And the maximum Ea,appfor HER at Ag(111)and Ag(110)in 0.5 mol/L H2SO4also appears at Ea,appclose to the PZC’s of Ag(111)and Ag(110)[12,23,24].The similar potential-dependent behavior of Ea,appand A for HER found at pc-Ag and pc-Ag(111)in different labs as well as its well correlation with the PZCs indicate that such phenomenon should be a well reproducible behavior for such system.
B.Kinetic implications from the temperature effects on HER
The widely accepted mechanism for HER at Ag electrode in acidic electrolyte is that HER goes through Volmer-Heyrovsky pathway with adsorbed Hadatom as reaction intermediate,and the f i rst charge transfer step (Eq.(2))is the rate determining step(rds),which is f i rst order with respect to H+.The total current density for HER can be written as:
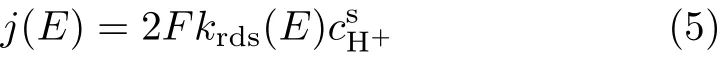
where 2 is the number of electrons transferred for formation of one H2molecule,F is the Faraday constant,is the concentration of H+near the electrode surface. krds(E)is the rate constant for the Volmer reaction, which can be expressed as:

where A(η)and Ea(η)are the pre-exponential factor and the activation energy for the Volmer reaction at overpotential η for HER.According to the transition state theory(TST),for elementary electrochemical reaction,the activation energy is just the Gibbs free energy change(denoted as∆6=Gact)from reactants to the transition state at reaction potential of E.It can be expressed as

For Volmer reaction,GRandare the Gibbs free energy of the hydrated proton in the reactant and transition state,the term βFη represents the contribution from potential induced change in the free energy of the electron.For HER at constant temperature and pressure,is the sum of the change in internal energy from reactants to the transition states∆,the related pressure volume change Pand temperature-entropy change T∆6=Sactterms,which can be expressed as:
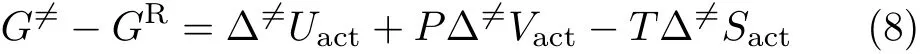
Since the reaction occurs at the electrode/electrolyte interface,the contribution of P∆6=Vactmay be neglected, there is

Obviously,both∆6=U and∆6=S are potential dependent.Substituting Eq.(9)into Eq.(6),and by proper rearrangement,we have
where A0represents the part of pre-exponential factor which is potential independent.Since the Volmer reaction is the rds for HER at Ag electrode and it is the fi rst step during the sequence for HER,and assuming that all reaction sites are uniform,+βFη for Volmer reaction should be proportional to the Ea,appfor HER derived from the experiments.In analog,for the Volmer reaction should be proportional to the experimentally determined A for HER,which ref l ects the effects of solvent dynamics[25]. For the Volmer reaction,it is expected that the reactants,i.e.,proton-containing complex(may be H9O4+or H5O2+or H3O+)to be discharged are mainly located in the outer Helmholtz layer[14],while the transition state may be in a region somewhat closer to the electrode surface.During the discharge process,the hydrated proton has to move close to the electrode surface, gets discharged somewhere near the electrode surface, replaces the water molecules adsorbed at the surface and f i nally forms adsorbed Had.
Since η is negative for HER,hence negative shift in electrode potential should lead to a decrease in the activation energy,i.e.,U6=-UR+βFη,this is opposite to what is observed for HER at Ag in the low overpotential region(e.g.,from-0.25 V to-0.4 V,Fig.4).Hence there must be an increase of the term of-URin this potential region,which overcompensates the decrease in activation energy induced by the increase in the potential energy of the electron.Since the binding energy for Ag-Hadis smaller than that for Ag-OH2[6,26],it is expected that a signif i cant part of activation energy has to be paid to get adsorbed water molecule detached from Ag surface,i.e.,to overcome the electrostatic interaction between water and the electrode surface as well as to break the hydrogen bond(HBs)of the adsorbed water molecule from its HBs network.
Since-0.4 V is just the PZC of pc-Ag in such system [23],in the potential region from-0.25 V to-0.4 V,the electrode surface has been positively charged,and the water molecules at the surface orient with their O atoms pointing toward electrode surface,where the HBs are partly broken.On the other hand,at PZC the water molecules form ice-like structure with well organized HB network.When decreasing from-0.25 V to-0.4 V, the positive electric field across the interface gradually decreases,HBs between the water molecules within the fi rst layer close to the electrode surface become stronger. And since the electric field is not so strong,in order to replace the water molecule from the surface the energy paid to break the HBs is higher than that for overcoming the electrostatic interaction.As a result,-URincreases from-0.25 V to-0.4 V.Alternatively,this may also be understood that from-0.25 V to-0.4 V during the Volmer reaction the entropy change for HB network environment increases.
On the other hand,compared to the reactant,the transition state is closer to the electrode surface and becomes restricted,the degree of freedom for the transition state is smaller than that for the reactants,hence-SRis negative.The increase of the preexponential factor for HER with negative shift of potential from-0.25 V to-0.4 V may be due to thatS| decreases in this low overpotential regime.With potential negative shift from-0.25 V to-0.4 V,the electric field across the interface decreases,hence the di ff erence between the structure of the transition state and that of the reactant becomes smaller.The decrease in|∆6=S| leads to a signif i cant increase in A0.At molecular level,the increase of A with potential from -0.25 V to-0.4 V may be envisaged by the increase in the transformation dynamics from the reactants’confi guration to the transition state with decrease in the positive electric field across the interface.This e ff ect prevails the adverse e ff ect from potential induced increase in activation energy and leads to a net increase in HER current at Ag from-0.25 V to 0.4 V.
At potentials more negative than the PZC of Ag,it is seen from Fig.4 that when the potential is negatively shifted from-0.4 V to-0.55 V,the Ea,appdecreases by ca.4 kJ/mol,from the Tafel plots given in Fig.5, it is found that the symmetric factor for the Volmer reaction is ca.0.3.Hence,the decrease in activation energy with electrode potential can be solely attributed to the term of βFη,while the contribution of the potential dependent change for the term U6=-URto the activation energy is negligible.Furthermore, we found that at E<PZC,the pre-exponential factor does not change with potential at all,this suggests that the term∆6=S=S6=-SRdoes not change with potential much.Both the lacking of the change of∆6=U and∆6=S can be easily explained by the fact that at E<PZC, proton-containing complex(may be H9O4+or H5O2+or H3O+)[2]to be discharged orient with H end toward the electrode surface,the structure of the transition state is quite similar to that for the reactant.As similar to the outer-sphere electrode reaction,the term of potential induced change of electron energy of βFη is the key factor which controls potential-dependent change of HER kinetics in this potential region,because with the increase in the negative electric f i eld across the interface toward more negative potentials,the electrons can tunnel into the solution further away from the surface, in the solution side more reactants further away from electrode surface may take part in the reaction[27].It should be mentioned that compared to that for HER at Au or Cd electrode in acidic electrolyte,the Ea,appfor HER just displays a monotonically decrease toward negative potentials,due to that the potential for HER is always negative of PZC of Au(ca.0.35 V)[28]and Cd electrodes.This is in good agreement with previous observation that at metals with low catalytic activity for HER,usually metals with more positive PZC display higher HER activity than those with lower PZCs [29].
It should be mentioned that Ea,appwe derived from this study as well as those obtained by Schmickler et al. at Ag(111)from temperature dependent activity measurements[12]are in the range of 20-30 kJ/mol,by taking the transfer coefficient of 0.5,the Ea,appat the equilibrium potential will be below 40 kJ/mol,which is smaller than∆6=G deduced from the recent theoretical calculation[13].For example,Schmickler et al.reported that at single crystalline Ag electrode,∆6=G is about 0.75 eV[11].On the other hand,from DFT calculation Norskov et al.have also found that at Pt(111),∆6=G for HER is ca.0.85 eV[30],which is much higher than the experimentally observed value of ca.0.2 eV by Markovic et al.[9].Such discrepancies indicate that either the models or the method used for theoretical calculation may not be appropriate.We hope such comparison may help theoreticians to construct more reliable model and develop more appropriate method in order to understand the essence for reactions such as electrochemical HER microscopically.
IV.CONCLUSION
Hydrogen evolution reaction at polycrystalline silver electrode in 0.1 mol/L HClO4solution is investigated by cyclic voltammetry in the temperature range of 278-333 K.A clear increase in HER current with reaction overpotential and temperature is observed. The apparent activation energy Ea,appincreases from 24 kJ/mol to 32 kJ/mol with electrode potential negative scan from-0.25 V to-0.4 V.And with further negative shift in electrode potential from-0.4 V to-0.55 V,Ea,appdecreases gradually to 28 kJ/mol. On the other hand,with negative potential shift from -0.25 V to-0.40 V the pre-exponential factor A also displays a monotonically increase,and it remains nearly unchanged at potentials from-0.4 V to-0.55 V.And -0.4 V is just near the PZC for pc-Ag.
The monotonical decrease in Ea,appwith electrode potential and the potential-independence of A at E<PZC agree well with the prediction from the Butler-Volmer’s law,suggesting that under such conditions, the reaction is similar to outer-shpere reactions.The increases in Ea,appand A with negative shift in electrode potential from the onset potential for HER to PZC is just opposite to what is predicted by Butler-Volmer equation.During the Volmer reaction,the displacement of adsorbed water leads to an increase in the change of both the internal energy and the entropy from reactants to the transition states.Present results reveal that the solvent dynamics and the related entropy term (pre-exponential factor)may contribute greatly to the kinetics for electrode reaction.
V.ACKNOWLEDGMENTS
This work was supported by the One Hundred Talents Program of the Chinese Academy of Science,the National Natural Science Foundation of China(No.21073176),and the National Basic Research Program of China National Science and Technology (No.2010CB923302).
[1]A.R.Despic,Comprehensive Treatise of Electrochemistry,New York and London:Plenum,(1983).
[2]F.Wilhelm,W.Schmickler,R.Nazmutdinov,and E. Spohr,Electrochim.Acta 56,10632(2011).
[3]B.E.Conway,Sci.Pro.71,479(1987).
[4]R.Parson,Catalysis in Electrochemistry:From Fundamentals to strategies for Fuel Cell,John Wiley&Son: Development,1(2011).
[5]S.Trasatti,J.Electroanal.Chem.39,163(1972).
[6]J.K.Norskov,T.Bligaard,A.Logadottir,J.R. Kitchin,J.G.Chen,and S.Pandelov,J.Electrochem. Soc.152,J23(2005).
[7]M.T.M.Koper,J.Solid State Electrochem.17,339 (2013).
[8]E.Skulason,V.Tripkovic,M.E.Bjorketun,S.Gudmundsdottir,G.Karlberg,J.Rossmeisl,T.Bligaard, H.Jonsson,and J.K.Norskov,J.Phys.Chem.C 114, 18182(2010).
[9]N.M.Markovic,B.N.Grgur,and P.N.Ross,J.Phys. Chem.B 101,5405(1997).
[10]Q.Zhang,Y.Liu,and S.Chen,J.Electroanal.Chem. 688,158(2013).
[11]E.Santos,P.Quaino,and W.Schmickler,Phys.Chem. Chem.Phys.14,11224(2012).
[12]D.Eberhardt,E.Santos,and W.Schmickler,J.Electroanal.Chem.461,76(1999).
[13]Z.Q.Tang,L.W.Liao,Y.L.Zheng,J.Kang,and Y. X.Chen,Chin.J.Chem.Phys.25,469(2012).
[14]E.Santos,P.Hindelang,P.Quaino,and W.Schmickler, Phys.Chem.Chem.Phys.13,6961(2011).
[15]J.K.Norskov,T.Bligaard,A.Logadottir,J.R. Kitchin,J.G.Chen,and S.Pandelov,J.Electrochem. Soc.152,J23(2005).
[16]J.K.Norskov,E.Skulasson,J.Rossmeisl,T.Bligaard, G.Karlberg,J.P.Greeley,and H.Jonsson,Abstracts of Papers of the American Chemical Society,Vol.233, Washington,DC 20036 USA:Am.Chem.Soc.,(2007).
[17]J.F.Li,Y.F Huang,S.Duan,R.Pang,D.Y.Wu,B. Ren,X.Xu,and Z.Q.Tian,Phys.Chem.Chem.Phys. 12,2493(2010).
[18]X.Xu,B.Ren,D.Y.Wu,H.Xian,X.Li,P.Shi,and Z.Q.Tian,Surf.Interf.Anal.28,111(1999).
[19]D.Y.Wu,S.Duan,X.M.Liu,Y.C.Xu,Y.X.Jiang, B.Ren,X.Xu,S.H.Lin,and Z.Q.Tian,J.Phys. Chem.A 112,1313(2008).
[20]F.Yang,Master Thesis Dissertation,Heifei:University of Science and Technology of China,(2013).
[21]J.Kang,Master Thesis Dissertation,Heifei:University of Science and Technology of China,(2013).
[22]Q.J.Chen,Y.L.Zheng,L.W.Liao,J.Kang,and Y. X.Chen,Sci.Sin.Chim.41,1777(2011).
[23]K.A.Soliman and L.A.Kibler,Electrochim.Acta 52, 5654(2007).
[24]W.Schmickler and E.Santos,Interfacial Electrochemistry,Springer-Verlag Berlin and Heidelberg GmbH& Co.K,(2010).
[25]S.Hammes-Schiffer and A.A.Stuchebrukhov,Chem. Rev.110,6939(2010).
[26]X.D.Song,Y.F.Zhao,P.X.Zhang,and G.H.Zhang, Inter.J.Quantum Chem.111,2109(2011).
[27]R.Pang,L.J.Yu,D.Y.Wu,B.W.Mao,and Z.Q. Tian,Electrochim.Acta 101,272(2013).
[28]D.Eberhardt,E.Santos,and W.Schmickler,J.Electroanal.Chem.419,23(1996).
[29]L.M.Doubova,and S.Trasatti,J.Electroanaly.Chem. 467,164(1999).
[30]E.Skulason,G.Karlberg,J.Rossmeisl,T.Bligaard,J. P.Greeley,H.Jonsson,and J.K.Norskov,Abstracts of Papers of the American Chemical Society,Vol.233, Washington,DC 20036 USA:Am.Chem.Soc.,(2007).
ceived on May 28,2013;Accepted on June 8,2013)
∗Author to whom correspondence should be addressed.E-mail:yachen@ustc.edu.cn,Tel./FAX:+86-551-6360035
 CHINESE JOURNAL OF CHEMICAL PHYSICS2014年1期
CHINESE JOURNAL OF CHEMICAL PHYSICS2014年1期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Experimental and Theoretical Investigation on Excited State Intramolecular Proton Transfer Coupled Charge Transfer Reaction of Baicalein
- Theoretical Study of Reagent Rotational Excitation effect on the Stereodynamics of H+LiF→HF+Li Reaction
- Infrared Spectroscopy of COIsolated in Solid Nitrogen Matrix
- effects of Rotational Isomerism and Bond Length Alternation on Optical Spectra of FTC Chromophore in Solution
- High Pressure Structural Instability and Thermal Properties of Rutile TiO2from First-principles
- Anisotropy of Thermal-expansion for β-Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine:Quantum Chemistry Calculation and Molecular Dynamics Simulation