
家族性多发性内分泌腺瘤2A型的基因诊断
2014-07-18 12:09:39朱宏建
武警医学 2014年7期
丁 维,朱宏建
家族性多发性内分泌腺瘤2A型的基因诊断
丁 维1,2,朱宏建1,2
目的提高对多发性内分泌腺瘤2型(multiple endocrine neoplasia 2, MEN2)的诊断和预测诊断水平。方法对临床已诊断首发症状为嗜铬细胞瘤的一个家族内两姐妹患者进行RET(外显子10~16)、VHL( 外显子1~3)、 SDHD(外显子1~4)、SDHB(外显子1~8) 及SDHC(外显子1~6) 基因测序。发现两患者均有RET基因634号核酸发生突变,并对其家族其他成员的RET基因进行测序。结果其家族内共有5例患者均有RET基因634号突变(TGC 突变为 CGC),导致其氨基酸编码由半胱氨酸变为精氨酸。其中2例先为临床诊断然后基因诊断,1例先由基因诊断然后临床诊断,2例(均为儿童,分别为6岁及7岁)为基因诊断,目前尚未发病。其中3例行腹腔镜双侧肾上腺肿瘤切除术,病理确诊为双侧嗜铬细胞瘤;1例行甲状腺切除术,病理诊断为甲状腺髓样癌。结论对于多发性内分泌多发肿瘤2A型患者及其家族成员行基因检查不仅可以作为确诊手段,更可以作为无症状者的预测诊断途径。
家族性多发性内分泌腺瘤2A型;甲状腺髓样癌;RET基因;基因测序
笔者于2013-03-27和2013-04-11临床发现2例双侧肾上腺嗜铬细胞瘤患者,且两人为亲姐妹。通过聚合酶链反应(polymerase chain reaction,PCR)和DNA测序,分析了其家族中6个成员RET基因的11号外显子突变情况,并进行了相应的临床处置。
1 对象与方法
1.1 对象
1.1.1 病例1 女性(该家庭中的3号成员),28岁,主因阵发性高血压及检查发现双侧肾上腺占位入院。体检:血压120/80~200/100 mmHg。颈部可扪及包块,质硬。血清降钙素正常。血清癌胚抗原32.75 ng/ml(正常值<5 ng/ml),血浆总皮质醇48.49 nmmol/L (240~618 nmol/L)。CT及MRI提示双侧肾上腺大小约6 cm×7 cm肿物。生长抑素受体显像提示双侧肾上腺生长抑素受体高表达。甲状腺超声提示双侧甲状腺肿物,左侧1.5 cm×2.8 cm×1.8 cm,右侧0.5 cm×0.5 cm×0.8 cm。2012-04-22于全麻下行腹腔镜双侧肾上腺肿物切除术,病理提示双侧嗜铬细胞瘤。术后血压恢复正常。于2012-09-05于全麻下行双侧甲状腺切除术,病理提示甲状腺髓样癌。
1.1.2 病例2 女性(该家庭中的4号成员),29岁,主因阵发性高血压半年,检查发现双侧肾上腺巨大肿物入院。尿去甲肾上腺素54.77 ng/24 h(正常值16.69~40.65 ng/24 h),尿肾上腺素4.56 ng/24 h(1.74~6.42 ng/24 h),尿多巴胺155.19 ng/24 h (120.93 ~330.59 ng/24 h)。影像学检查,MRI提示双侧肾上腺占位,左侧4 cm×3.5 cm×3 cm,右侧6.5 cm×6 cm×5.5 cm。于2012-04-26于全麻下行双侧腹腔镜肾上腺肿物切除术。病理提示嗜铬细胞瘤。 术后血压恢复正常。笔者对其家族成员进行基因测序并绘制家族系谱图(图1)。
1.2 基因测序方法 2013-06-08抽取2个原发患者的外周血(指南不推荐肿瘤基因测序)[1]测定RET(外显子10 ~16)、VHL( 外显子1~3)、SDHD(外显子1~4)、SDHB(外显子1~8) 及SDHC(外显子1~6) 基因。通过PCR及毛细管电泳和荧光标记技术的DNA测序发现2个原发患者在RET基因634号位点有相同的突变,导致半胱氨酸变为精氨酸。之后测定其余家庭成员RET基因11号外显子的序列。
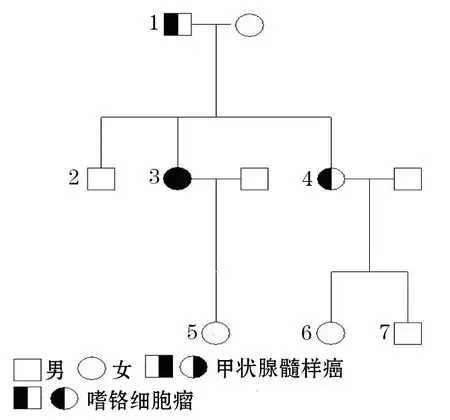
图1 MEN2A家庭系谱
1.2.1 反应引物 PCR放大RET、VHL、SDHD、 SDHB 及SDHC 。RET基因外显子所用引物见表1。

表1 MEN2A患者RET基因PCR中的引物
1.2.2 实验操作 (1)PCR扩增体系:包括GC buffer I 12.5 μl,dNTP(2.5 mmol)2 μl,上游引物(10 mmol)1 μl,下游引物(10 mmol)1 μl,DNA 2 μl,r Taq(Takara) 0.2 μl,水6.3 μl,总共25 μl。(2)PCR反应程序:DNA在DYY-8C电泳仪(北京六一仪器厂)电泳后,于PTC-200 PCR仪(BIO-RAD)98℃变性5 min,55℃退火20 s,72℃延伸30 s。此循环共循环30次。(3)PCR产物纯化:向96纯化板(Millipore)孔中加入100 μl ddH2O,取出PCR产物至96纯化板中室温静置10 min,真空抽滤泵抽(Millipore)10 min至抽干,向96孔纯化板中加入40 μl ddH2O,室温溶解10 min,取出对应的孔中的纯化产物至另一96孔PCR板中。(4)PCR产物测序:测序体系包括DNA 3 μl(30~50 ng),引物(3 pM)1 μl,Bigdye 0.5 μl,ddH2O 0.5 μl,总体积5 μl。 (5)PCR测序反应程序:95 ℃, 2 mins → (95℃ 10 s → 51 ℃ 10 s → 60 ℃190 s) × 25个循环 → 12℃保温,最后用UV-IV紫外分析仪(北京市新技术应用研究所)测出基因序列。
2 结 果
基因测序结果见图2,家族中基因突变情况见表2。发现其家族内有5个成员均有RET基因634号突变(TGC 突变为 CGC),导致其氨基酸编码由半胱氨酸变为精氨酸。其中2例先临床诊断然后基因诊断(即前述姐妹俩);1例先是基因诊断然后临床诊断(该家庭中的1号成员;该患者后在我院进行了手术,术后病理证实为双侧肾上腺嗜铬细胞瘤);2例(均为儿童,分别为6岁、7岁)为基因诊断,目前尚未发病。

表2 NEN2A家庭成员中RET基因突变情况
注:该家庭中的2号成员未到院检测
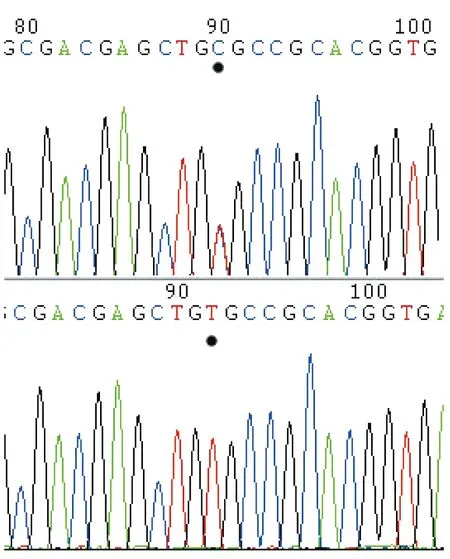
图2 RET基因634号位点突变(T→C),突变处用黑点标记出
3 讨 论
3.1 多发性内分泌腺瘤2型临床表现 多发性内分泌腺瘤2型(multiple endocrine neoplasia 2, MEN2)可以分为三类:MEN 2A、家族型甲状腺髓样癌(familial medullary thyroid carcinoma,FMTC)和 MEN 2B。MEN 2A 临床诊断需要在一个患者身上发现2个或2个以上特定的内分泌肿瘤(甲状腺髓样癌、嗜铬细胞瘤或者甲状旁腺肿瘤)。在MEN2A患者中有95%出现甲状腺髓样癌,50%出现嗜铬细胞瘤,20%~30%出现甲状旁腺功能亢进[2,3]。MNE2B比MEN2A更有侵略性,表现为甲状腺髓样癌、嗜铬细胞瘤、马方综合征和黏膜肿瘤[4,5]。
3.2 RET基因遗传学特点 RET基因位于10号染色体长臂11.2,编码一种络氨酸激酶受体[6]。此受体对神经脊细胞、泌尿生殖系统、中枢及周围神经系统发育起重要作用。RET基因突变是唯一已知导致MEN2型的基因。分子遗传学检测发现RET基因突变在MEN2A中占98%,在MEN2B中超过98%,在FMTC中占大约95%[7,8]。一些突变可以激活RET激酶活性,导致癌症的发生。MEN2A的发生大多数由于RET基因细胞外区域氨基酸置换引起[9,10]。对于嗜铬细胞瘤和非家族型嗜铬细胞瘤可能存在可遗传的RET、 VHL、 SDHD、 SDHB、 TMEM127、 MAX 及SDHA基因突变。
3.3 基因检测的优越性 MEN2是少数几个需要进行基因检测的疾病,MEN97研讨会认为,在行甲状腺切除术前需要进行RET基因突变检测,而不是降钙素刺激试验[2]。理由如下:(1)早期发现和早期介入可以改变甲状腺髓样癌临床进程。通过降钙素刺激实验筛查MTC患者并在其青少年期进行手术治疗可以治愈大部分患者。(2) 早期MTC患者即使是大多数婴儿也可以耐受甲状腺切除术。(3)RET基因突变的研究表明,用降钙素水平异常这一指标来决定是否进行甲状腺切除术有5%~10%的假阳性率。(4)RET基因检测相比降钙素实验而言有更高的确诊率和更低的漏诊率及假阳性。
大约98%的MEN2病例有明确的RET基因突变,现发现与MEN2有关的RET基因突变主要集中在外显子10、11、13、14、15和16。因此,这些外显子需要优先检测是否存在突变。如果未发现突变,则需要检测剩余的15个外显子。可遗传的嗜铬细胞瘤包括MEN2A、MEN2B、 Von-Hippel-Lindau综合征(VHL)、多发性神经纤维瘤1型、副神经节瘤和遗传型嗜铬细胞瘤。嗜铬细胞瘤散发病例发病率5%~13%。在这些病例中进行RET、VHL及NF1基因检测分析可能发现异常突变。
3.4 RET基因检测的临床意义 RET基因突变位点与MEN2疾病变化有对应关系,包括侵略性MTC。因此,在进行甲状腺监测时需要特别注意RET基因突变位点和其家庭成员特征性临床表现。对于MTC的危险度需根据RET基因突变部位分层对待及处理。MEN2B或者有RET883、918或922密码子基因突变的小孩,危险度为3级,需要在其6个月大时行甲状腺切除术。在RET611、618、620或者634密码子突变的小孩,危险度为2级,需在5岁前行甲状腺切除术。在密码子634突变的病例中,早期甲状腺切除术已经发现2岁小孩的微小MTC病变及5岁小孩的淋巴结转移。一般来说,这些位点突变的病例MTC生长比较缓慢。一些学者建议在5岁时行甲状腺切除术,另一些则建议10岁进行[11]。
通过基因测序,一些MEN2携带者在发展为MTC前就进行了甲状腺切除术。但是,血清降钙素及降钙素刺激实验仍然是术前及术后筛查及检测肿瘤的理想指标。
嗜铬细胞瘤已经在除外RET基因609、768、val804met及891密码子的其他所有突变中均有发现。在634密码子突变中,最早可在5~10岁发现嗜铬细胞瘤。对于更高危险度的突变,对于嗜铬细胞瘤的检测需在行甲状腺切除术时或者5~7岁时,并且需要每年检测一次。而对于RET基因609、768、val804met及891密码子突变者,检测可推迟一些。对于在634密码子突变导致氨基酸改变,患者更倾向发展为甲状旁腺功能亢进。突变在609、611、618、620、790及791密码子的患者则较少地发展为甲状旁腺功能亢进。血清甲状旁腺激素及钙水平需要每2~3年检测一次[12]。
3.5 产前咨询 由于基因检测可以准确地检测出是否患有MEN2疾病,因此对于MEN2致病基因携带者行产前咨询是有必要的。对于MEN2A家族成员,RET基因测序可以做到早期诊断,并对其进行早期干预处理。
[1] Brandi M L, Robert F. Gage L,etal. Guidelines for Diagnosis and Therapy of MEN Type 1 and Type 2[J]. J CLIN ENDOCR METAB, 2001,86(12):5658-5671.
[2] Steiner A L, Goodman A D.Powers SR Study of a kindred with pheochromocytoma,medullary carcinoma, hyperparathyroidism and Cushing’s disease: multiple endocrine neoplasia, type 2[J].Medicine, 1968, 47(5):371-409.
[3] Schuffeneker I, Virally-Monod M, Brohet R.Risk and penetrance of primary hyperparathyroidism in multiple endocrine neoplasia type 2A families with mutations at codon 634 of the RET proto-oncogene[J].J Clin Endocrinol Metab, 1998, 83(2):487-491.
[4] Williams E D, Pollock D J.Multiple mucosal neuromata with endocrine tumours: a syndrome allied to Von Recklinghausen’s disease[J].J Pathol Bacteriol, 1966, 91(1):71-80.
[5] Carney J A, Go V L, Sizemore G W.Alimentary-tract ganglioneuromatosis. A major component of the syndrome of multiple endocrine neoplasia, type 2b[J].N Engl J Med, 1976, 295(23):1287-1291.
[6] Ceccherine I, Romei C, Barone V,etal. Identification of the Cys634y Tyr mutation of the RET proto-oncogene in a pedigree with multiple endocrine neoplasia type 2A and localized cutaneous Lichen amyloidosis[J].J Endocrinol Invest, 1994, 17(3):201-204.
[7] Berndt L, Reuter M, Saller B,etal.A new hot spot for mutations in theret proto-oncogene causing familial medullary thyroid carcinoma and multiple endocrine neoplasia type 2A[J].J Clin Endocrinol Metab, 1998, 83(3):770-774.
[8] Niccoli-Sire P, Murat A, Rohmer V,etal.Familial medullary thyroid carcinoma with noncysteine RET mutations: phenotype-genotype relationship in a large series of patients[J].J Clin Endocrinol Metab, 2001, 86(8):3746-3753.
[9] Donis-Keller H, Dou S, Chi D,etal.Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC[J].Hum Mol Genet, 1993, 2(7):851-856.
[10] Mulligan L M, Kwok J B, Healey C S,etal.Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A[J].Nature, 1993, 363(6428):458-460.
[11] Brandi M L, Robert F,Gagel A A,etal.Guidelines for Diagnosis and Therapy of MEN Type 1and Type 2[J].J Clin Endocrinol Metab,2001, 86(12):5658-5671.
[12] Gagel R F, Tashjian A H, Cummings T,etal.The clinical outcome of prospective screening for multiple endocrine neoplasia type 2a: an 18-year experience [J].N Engl J Med, 1988, 318(8):478-484.
(2014-02-12收稿 2014-04-20修回)
(责任编辑 梁秋野)
AChineseMEN2ApedigreeassociatedwithmutationofRETproto-oncogene
DING Wei1,2and ZHU Hongjian1,2. 1. Clinical Institute of Chinese People’s Armed Police General Hospital of Medical College,Anhui University,Beijing 100039,China; 2.Department of Urology,General Hospital of Chinese People’s Armed Police Forces,Beijing 100039,China
ObjectiveBy gene sequence ,biochemical and imaging testing, we introduce the diagnosis ,especially the predictive diagnosis of MEN2a.MethodsPeripheral blood from 2 patients who were sisters presenting pheochromocytoma as the first manifestation of disease was tested for mutations of RET(exon 10 ,11, 12,13,14,15,16), VHL( exon 1 to 3), SDHD(exon 1 to 4), SDHB(exon 1 to 8) and SDHC(exon 1 to 6) by means of genomic polymerase chain reaction (PCR) amplification and DNA sequencing. The mutation affected RET proto-oncogene codon 634 and caused a cysteine to arginine substitution. Then we sequenced the other family members’ RET gene exon 11.ResultsWe have detected point mutation in 5 MEN 2A patients in a MEN 2A family at codon 634 (TGC to CGC) in RET exon 11 which caused a cysteine to arginine substitution. Three of them were diagnosed as having bilateral adrenal pheochromocytomas by pathological examination and bilateral tumor in thyroid gland by ultrasound study. One patient underwent bilateral thyroidectomy and pathological examination showed medullary thyroid carcinoma.ConclusionsThe identification of a DNA alteration in the MEN2A gene will permit predictive diagnosis. Molecular testing of individuals at risk in these MEN 2A families is considered part of the standard management for at-risk family members.
multiple endocrine neoplasia type2; medullary thyroid carcinoma; RET gene;gene sequence
武警总医院院级基金(WZ2012035)
丁 维,硕士研究生,E-mail:aydsummy@qq.com
1. 100039北京,安徽医科大学武警总医院临床学院;2. 100039北京,武警总医院泌尿外科
朱宏建,E-mail: hjzhu99@sina.com
R596.2
猜你喜欢
天津医科大学学报(2021年2期)2021-03-29 05:30:58
生物学通报(2020年11期)2020-10-22 01:20:20
临床肝胆病杂志(2020年9期)2020-09-28 07:48:02
中国临床医学影像杂志(2019年5期)2019-08-27 02:47:46
现代泌尿生殖肿瘤杂志(2018年5期)2018-11-29 06:10:02
中成药(2018年7期)2018-08-04 06:04:10
西南国防医药(2016年6期)2016-12-01 06:01:15
分子影像学杂志(2015年3期)2015-12-04 03:29:01
生命科学研究(2014年1期)2014-04-29 00:44:03
中国烟草学报(2012年2期)2012-04-09 06:44:56
