
紫衫醇C13侧链的微观结构及光谱性质
2014-07-13 03:39刘信平聂光华
原子与分子物理学报 2014年2期
刘信平,聂光华,王 艳
(湖北民族学院化学与环境科学学院湖北省湖北民族学院生物资源保护与利用重点实验室,恩施,445000)
1 引 言
抗癌特效药紫杉醇因资源严重短缺、水溶性太差,临床中存在诸多弊端,拓宽资源供应、寻找高效低毒的新型紫杉醇类拟药刻不容缓,化学半合成法成为当前乃至今后相当长时间内紫杉醇类拟药研发热点,而对紫杉醇C13侧链进行结构修饰改造是当前半合成研发的重点课题之一.如Ojima研究组合成了对C-3’修饰的奥兰索已进入临床试验阶段[1].Sang-H.L 等 用SH 取 代C-2’位[2],Gunda I.Georg 等用呋喃和吡啶取代C-3’位的Ph[3];目前的半合成实验研究报道相对较多,但从微观层面研究其结构性能尚未见报道.为了更好促进对C13侧链结构改造、修饰,得到有效的紫杉醇类拟药,了解其分子离子的微观结构及其性能是必要的.本文用量子力学理论计算方法[4,5],得到了C13侧链分子离子的微观结构及性能、前线分子轨道特征,并认证了其IR 光谱图,为更进一步的结构修饰改造提供必要的理论依据和实验基础.
2 计算方法
本文全部计算用Gaussian09 程序包,采取B3LYP方法,6-31+G*水平基组对所选体系进行了几何全优化、NBO 计算、频率计算等,收敛精度取程序内定值.为讨论方便,用M 代表C13侧链.
3 结果与分析
3.1 几何结构优化及自然原子电荷分析
用不同的初始构型对所选体系结构进行优化及频率计算,振动分析表明,所得的优化构型对应势能面上能量最小点(即无虚频振动频率),Mn(n=0,1,-1)体系都具有一定的稳定性,且稳定平衡结构基态的电子状态为:M 是2A,M+为:3A,M-为:1A,结构图及其相关结构参数如图1~3和表1所示.

图1 M(2 A)优化结构图Fig.1 The optimized structures of M(2 A)

图2 M+(3 A)优化结构图Fig.2 The optimized structures of M+(3 A)

图3 M-(1 A)优化结构图Fig.3 The optimized structures of M-(1 A)
由表1可知,Mn(n=0,1,-1)稳定构型的能量EM-< EM< EM+,因此稳定性顺序为:M-(1A)>M(2A)>M+(3A),根据偶极矩数值得到 它 们 极 性 强 弱 是M+(3A)> M-(1A)>M(2A).在空 间 构 型 上,M+(3A)的R(C1-C2)=0.5073nm 显著大于M(2A)的0.1734nm,与C1、C2相连的其它原子间的平衡几何R 都有一定程度的缩短,相应的键角和二面角计算结果表明:A(C1,C2,C4):M(2A)的=114.0859o、M+(3A)的=69.5679°,A(C2,C1,O24):M(2A)的=107.2020°、M+(3A)的=14.4680°,D(22,1,2,20):M(2A)的=41.5480°、M+(3A)的=137.6445°,D(24,1,2,4):M(2A)的=161.0554°、M+(3A)的=-117.1791°,D(4,18,25,26):M(2A)的=-152.7716°、M+(3A)的=8.7640°,M+(3A)空间构型随着C1-C2拉长的同时羧基以C2为轴点向C2-C4连线靠近,使得相应的键角、二面角发生较大的翻转.相对来说M-(1A)的空间构型变化程度要缓和的多,从结构图也可清晰看出.表2给出了化合物部分原子的自然原子电荷数.

表1 Mn(n=0,1,-1)微观结构性质 Table 1 calculated results of micro-properties and structures for Mn(n=0,1,-1)

表2 部分原子的净电荷布局 Table 2 The charge distributions of part atomic of Mn(n=0,1,-1)
由表2可知:M(2A)荷正电荷的主要原子为C1=0.5179、C2=0.7116、C4=0.3496、C25=1.274,荷负电荷的主要原子N18=-0.5767、O20=-0.3565、O22=-0.4246、O24=-0.4608、O26=-0.8531,从荷电数值可知,在中性分子中C1、C2、C25三个原子受亲核试剂进攻的可能性较大,难易顺序为C25>C2>C1,N18、O20、O22、O24是亲电试剂进攻的原子,难易顺序为N18>O24>O22>O20,即这7个原子为该物质化学活动部位;
M+(3A):C1=1.1214、C2=0.3486、C4=0.3690、C25=1.2730,荷负电荷的主要原子N18=-0.1534、O20=0.2302、O22=-0.2048、O24=-0.8642、O26=-0.8691,因此两个羰基的C1和C25是该阳离子主要正电荷原子,极易与亲核试剂结合成键.C2上的羟基O20荷正电荷,而C1上的羟基O22荷负电降低但羰基O24升高,即C1、C2上两个羟基的电子明显流向羰基O24.从酰胺及与之链接的苯环电荷分布可以看出,氨基、酰基,苯环三者出现一定程度的共轭,电荷从氨基向酰基、苯环偏移,使得N18上荷负电减少而苯环、酰基O26增加;
M-(1A):C1=-0.0373、C2=0.5209、C4=-0.5259、C25=0.0440,荷负电荷的主要原子N18=0.1816、O20=-0.1431、O22=-0.2482、O24=-0.6711、O26=-0.4133,从这7个原子电荷布局可知,整个分子电荷由苯酰胺一侧向另一端的C1、C2及与其相链接得羟基羧基转移,结果使得氨基N18反而荷正电而C1、C4却荷负电.
3.2 前线分子轨道特征分析
量子化学的前线轨道理论认为[6-8],分子参与反应分子轨道要发生变化,优先起作用的是前线轨道,最低空轨道与最高占据轨道的能量差△E(ELUMO-EHOM0)是分子稳定性的重要指标,其差值越大,分子稳定性越强;反之,分子越不稳定,越易参与化学反应,因此研究前线轨道的性质可以为确定活性部位以及探讨作用机制等提供重要信息.根据所选基组及分子离子体系基态稳定构型,NBO 计算得到Mn(n=0,1,-1)分子轨道信息见表3 所示.由表3 可知,M-(1A)的△E 最大,M+(3A)的最小,即稳定性顺序是M-(1A)>M(2A)>M+(3A),结果与前面分析一致.
计算得到Mn(n=0,1,-1)体系的前线分子轨道的电子云如图4~9所示.

表3 Mn(n=0,1,-1)的分子轨道特性Table 3 The properties parameters of the frontier molecular for Mn(n=0,1,-1)
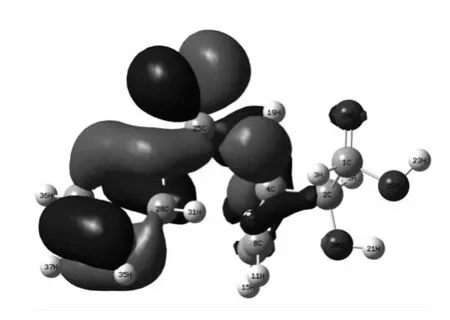
图4 M(2 A)的HOMO 电子云Fig.4 The electronic cloud of the HOMO for M(2 A)

图5 M(2 A)的LUMO 电子云Fig.5 The electronic cloud of the LUMO for M(2 A)
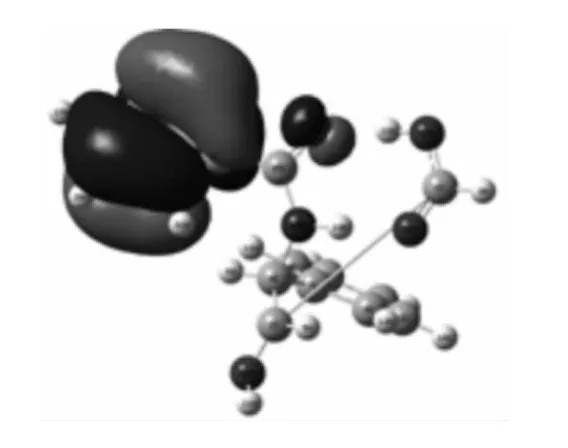
图6 M+(3 A)的HOMO 电子云Fig.6 The electronic cloud of the HOMO for M+(3 A)
由上图可知,M(2A)的HOMO 电子云主要集中苯酰胺一端,LUMO 电子云主要集中在另一端的羰基及羧基上,既苯酰胺端是反应时亲电试剂进攻部位,而另一端是亲核试剂进攻部位.M+(3A)的HOMO 电子云主要与C3链接的苯环及酰

图7 M+(3 A)的LUMO 电子云Fig.7 The electronic cloud of the LUMO for M+(3 A)

图8 M-(1 A)的HOMO 电子云Fig.8 The electronic cloud of the HOMO for M-(1 A)
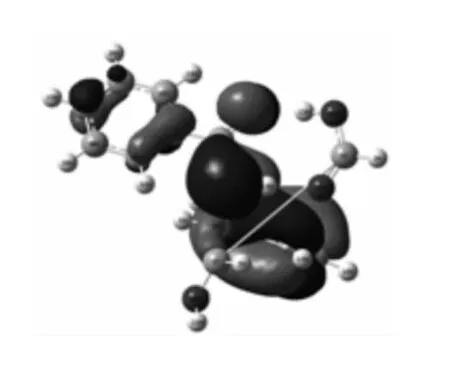
图9 M-(1 A)的LUMO 电子云Fig.9 The electronic cloud of the LUMO for M-(1 A)
胺基团部位,LUMO 主要集中在苯酰胺基团部位,M-(1A)的HOMO 电子云主要与C3-C2所链接的羰基及两个羟基部位,LUMO 电子云在苯酰胺基团部位.由此可知,Mn(n=0,1,-1)体系化学活性部位主要是苯酰胺基团部位及C3-C2所链接的羧基和羟基位置.由此可证明Mn(n=0,1,-1)均属两性反应体系,即可是电子供体也可是接受体,根据这一特性可以在设计目标物结构改造时有针对性地选择反应试剂,达到预期目的.
3.3 电子吸收IR光谱分析
频率计算得到其IR 吸收光谱数据,并通过该计算方法和基组的校正因子校正后的IR 图谱如图10所示,对各峰的振动模式进行了归属,谱进行了详细的指认.
4 结 论
本工作在B3LYP/6-311+G*水平基组上对Mn(n=0,1,-1)体系基态进行了结构优化、频率、自然键原子轨道计算,结果表明:分子、正负离子稳定结构的组态是2A、3A、1A,稳定性顺序为:M-(1A)>M(2A)>M+(3A),极性强弱是M+(3A)>M-(1A)>M(2A),M+(3A)的空间构型与M(2A)相比在一些部位变化大,特别是在C1-C2位置,键长拉长很多而键角减小很多.净电荷、分子轨道特征分析一致确定分子、正负离子体系的酰胺、羟基、羧酸基团是参加化学反应时的活性部位,能与亲核或亲电试剂反应,是关键药效团的活性部位,这一结论与紫衫醇实验研究的构效关系结论一致.得到了C13侧链分子红外光谱,并对谱图中各峰进行了振动模式归属及指认.本研究成果为进一步研究紫衫醇C13侧链的修饰改造,合成更多高效低毒的紫衫醇类拟物提供可参考的理论基础和实验依据,具有很大的理论意义和应用价值.

图10 C13 侧链子IR 谱图(振动频率:cm-1,红外强度:Km/mol)Fig.10 IR spcctra of the taxol C13side chain(Frequcncy:cm-1,intensity:Km/mol)
[1] Ojima I,Duclos O,Kuduk S,et al.Synthesis and biological activitv of 3’-alkyl-and 3’-alkenyl-3’-de-phenyldocetaxels[J].Bioorg Med Chem.Lett.,1994,4(21):2631.
[2] Sang H L,Xin Q,Ju Y Y.Preparation ofβ-amino-αmercapto acids and amides:stereocontrolled syntheses of 2’-sulfur analogues of the taxol C-13 side chain,both syn and anti S-acetyl-N-benzoyl-3-phenylisocysteine[J].Original Research Article Tetrahedron,2002,58(14):2777.
[3] Gunda I G,Gerlandine C B.Heteroaromatic taxol analogues:the chemistry and biological activities of 3’-furyl and 3’-pyridyl substituted taxanes[J].Original Research Article Bioorganic & Medicinal Chemistry Letters,1994,11(4):1381.
[4] Liu X P,ZHang C.Potentional energy function and micro-properties of ground SeFn(n=0,+1,+2)and extited B12Πand B22Π[J].J.At.Mol.Phys.,2008,25(1):183(in Chinese)[刘信平,张驰.SeFn(n=0,+1,+2)基态及SeF低激发态的势能函数与微观性质[J].原子与分子物理学报,2008,25(1):183]
[5] Lei X L,Zhu H J,Wang X M.Study on structures and properties of ZrnB(n=1~13)clusters using DFT[J].Acta Phys.Chim.Sin.,2008,24(9):1655(in Chinese)[雷雪玲,祝恒江,王先明.用密度泛函理论研究ZrnB(n=1~13)团簇的结构及性质[J].物理化学学报,2008,24(9):1655]
[6] Hehre W J,Radom L,SebleyerPv R,et al.Abinifio molecular orbital theory[M].New York:John Wiley&Sons.Ine,1986.
[7] Tang O Q,Yang Z Z,Li Q S.Quantum chemistry[M].Bei Jing:Science Press,1982.(in Chinese)[唐敖庆,杨忠志,李前树.量子化学[M].北京:科学出版社,1982]
[8] Wang J F,Feng J K.Theoretical studies of the structure optimization,frontier orbitals and properties of bifluorene and Its derivatives[J].Chem.J.Chin.Universities,2008,29(1):177(in Chinese)[王继芬,封继康.二芴及其衍生物的结构优化、前线轨道及其性质的理论研究[J].高等学校化学学报,2008,29(1):177]
猜你喜欢
军民两用技术与产品(2022年1期)2022-06-01
山东农业大学学报(自然科学版)(2021年3期)2021-07-29
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
世界农药(2019年3期)2019-09-10
世界农药(2019年3期)2019-09-10
世界农药(2019年2期)2019-07-13
粘接(2017年4期)2017-04-25
北京航空航天大学学报(2017年10期)2017-04-20
原子与分子物理学报(2015年3期)2015-11-24
航天返回与遥感(2014年4期)2014-07-31
