
基于高通量测序的辽东栎转录组学研究
2014-07-12 05:42刘玉林李伟张志翔
生物技术通报 2014年7期
刘玉林 李伟 张志翔
(1.北京林业大学生物科学与技术学院,北京 100083;2.北京林业大学自然保护区学院,北京 100083)
随着煤、石油、天然气等传统能源的枯竭及燃烧所带来的生态环境的恶化,燃料乙醇作为新兴的生物质能源得到越来越多的国家的重视[1,2]。在许多相对发达的国家,生产燃料乙醇主要是以玉米、甜高粱等作物为原材料[3,4],而对于我国人均耕地面积处于世界平均耕地面积以下的现状,利用粮食生产燃料乙醇明显是不现实的[5]。因此,寻求廉价易得的“非粮”生物质能源的原材料已成为国内外学者竞相研究的热点。
栎树(橡树或柞树),为壳斗科栎属植物的统称。全世界约有300余种,我国约有60余种[6]。栎树种子富含淀粉,含量可达到50%-70%,而我国约有栎树林1 800万hm2,年产约1 000万t栎树种子,可生产燃料乙醇约250万t[7],具有巨大的开发潜力。若利用栎树种子发展燃料乙醇,不仅在不影响国家粮食安全的情况下发展燃料乙醇产业,同时又能加大栎树种子的利用率,刺激林业经济的发展[8,9]。
利用淀粉植物生产燃料乙醇,淀粉含量是一重要指标。目前,国内外对栎属植物中淀粉的研究仅停留在物理与化学性质的分析上[10,11],还未对栎属植物中淀粉合成代谢的分子机理进行深入的研究,进而利用分子手段进一步提高淀粉的含量和产量。虽然国外学者利用传统的测序方法已对夏栎(Quercus robur)和无梗花栎(Quercus petraea)进行一定层次的研究[12-14],但大部分研究和所获得的基因序列来自于芽和叶等组织,还未包括主要积累淀粉的果实中的转录数据,因而对栎树中淀粉合成代谢的研究有一定的局限性。随着高通量测序技术的发展,测序成本的降低,基于高通量测序技术的转录组分析越来越成为非模式植物中发掘功能基因的一种有效的手段。近年来,研究人员已对多种植物进行了转录组测序和基因的发掘与研究[15]。
本研究利用Illumina Solexa Hiseq 2000高通量测序技术对广泛分布于我国的陕西、宁夏、黑龙江、河北、河南、吉林、青海、辽宁、山西、四川、甘肃、内蒙古、山东等地及朝鲜地区的温带、暖温带森林植被的主要建群种,且种子中淀粉含量高达62.88%左右[16]的辽东栎的芽、花、叶和果实的混合样品进行转录组测序,旨在获得辽东栎更多的转录本和更为全面的转录组信息,发掘辽东栎中参与淀粉合成代谢的基因和潜在的SSR(Simple sequence Repeat)标记,为进一步研究辽东栎中淀粉的合成机制,选育优良树种及辽东栎乃至栎属植物的进化与多样性分析奠定分子基础。
1 材料与方法
1.1 材料
本研究中辽东栎的芽、花、叶与果(5个阶段:开花后20、40、60、80和100 d均采集)均采集于北京市门头沟区小龙门国家森林公园(E11526’,N3959’),每份样品至少取自10个单株。样品采集后立即放入液氮中,后存放于-80℃冰箱备用。
1.2 方法
1.2.1 总RNA的提取和Solexa测序 总RNA的提取采用RNeasy Plant Mini Kits(Qiagen,Inc.,Valencia,CA,USA)试剂盒按照实验手册操作步骤提取。来自芽、花、叶和果的等量RNA混合后,取10 μg用于cDNA文库构建,cDNA文库的构建参考文献[17] 的方法。利用Illumina Solexa Hiseq 2000的paired-end测序方法进行转录组测序。
1.2.2 序列的处理与拼接 鉴于Solexa数据错误率对结果的影响,对原始数据进行质量预处理。先利用滑动窗口法去除低质量片段:质量阈值20(错误率=1%),窗口大小5 bp,长度阈值35 bp;再切除reads中含N部分序列:长度阈值35 bp;最后利用软件Trinity对辽东栎有效reads进行从头拼接[18]。
1.2.3 功能注释 使用BLASTx程序将拼接所得的unigene与核酸、蛋白质序列数据库比对(E值<1e-5),并选取最佳注释。蛋白质数据库包括Swiss-Prot、GenBank非冗余蛋白数据库(Nr)、蛋白质直系同源簇数据库(Clusters of Orthologous Groups of proteins,COGs)、京都基因和基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)及Gene Ontology(GO)。其中,unigene通过COG、GO和KEGG数据库的分类的参考文献[19] 的方法。
1.2.4 SSR位点的筛选 利用MISA软件在所有unigene 中搜索SSR 位点,参数设置如下:二核苷酸至少重复次数为6,三核苷酸、四核苷酸、五核苷酸和六核苷酸至少重复次数均为5。
2 结果
2.1 Illumina Solexa 测序和序列拼接
采用 Illumina Solexa Hiseq 2000 测序技术对辽东栎的芽、花、叶和果实的混合样进行转录组测序,共获得46 400 862条原始序列,总长4.64 Gb。经过预处理,最终得到了有效序列40 621 588条,数据量为3.8 Gb,平均长度为94.95 bp。使用软件Trinity对有效reads进行从头拼接最终得到了151 339个长度大于200 bp的contig,总长度约为130.92 Mb,最大长度、平均长度以及N50分别为11 284、865和1 442 bp。取每个contig下最长的转录本作为unigene,得到了95 800个unigene,总长度约为73.57 Mb,平均长度与N50分别为768 bp和1 373 bp。其中,大于2 000 bp的序列共有7 975条,占unigene总数的8.32%(图1-A)。
2.2 序列的比对、功能注释及unigene的特征分析
将所获得的unigene与公共数据Nr和Swiss-Prot进行Blastx比对,通过gene的相似性进行功能注释,共有57 637条unigene获得了基因注释(hit),占总unigene总数的60.16%,而在其余未得到任何一个上述数据库的注释(no-hit)38 163条unigene(39.84%)中,有37 752条unigene(98.92%)小于或等于1 000 bp(图1-B)。
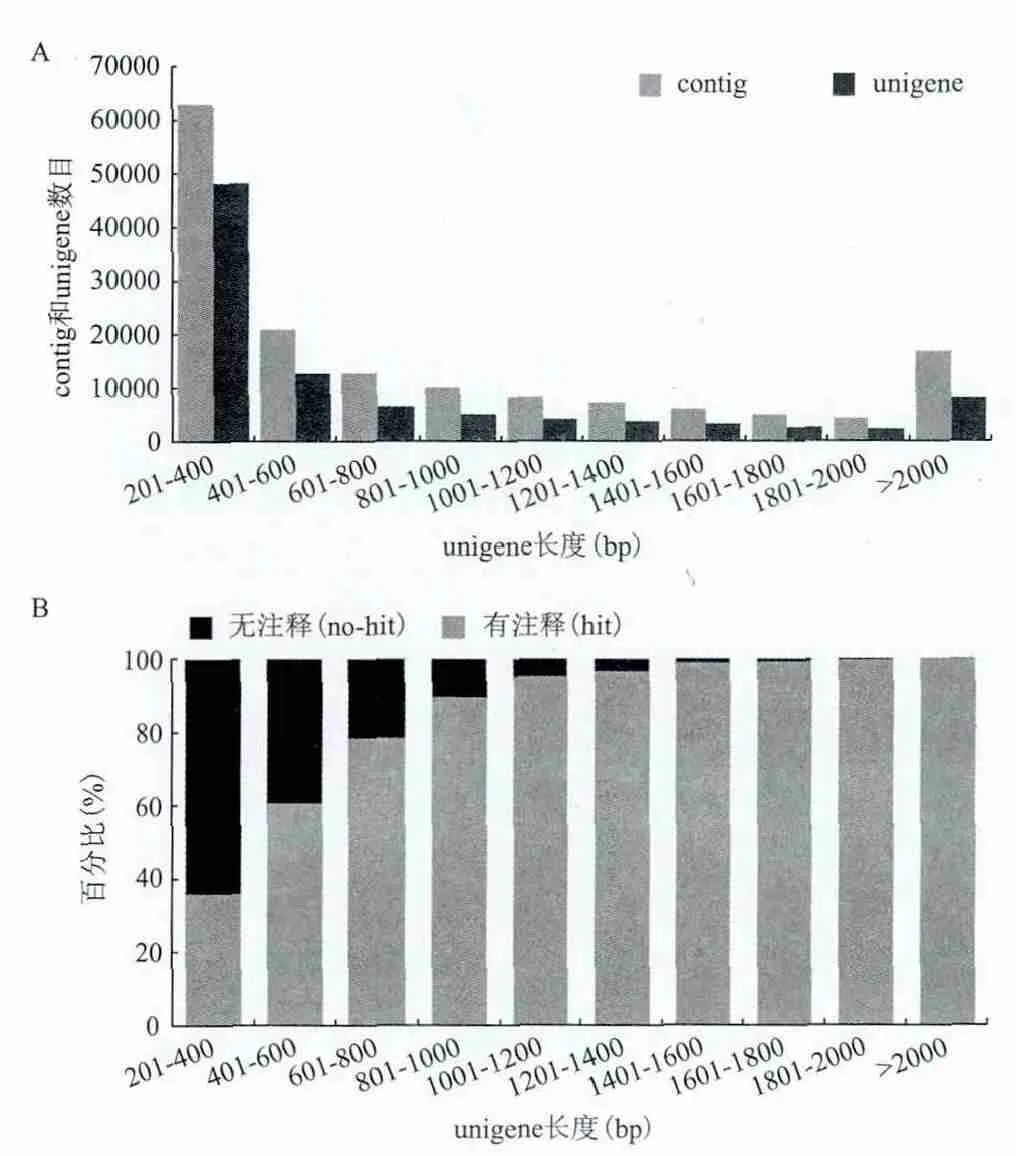
图1 contig和unigene的长度分布(A)及有注释和无注释的unigene的分布比例(B)
2.3 功能分类研究
为进一步研究辽东栎中unigene的功能分类,将所得到的95 800条unigene在COG和GO数据库中进行比对及功能注释与分类。
在COG分类中,共有36 407 条unigene(占unigene总数的38%)被注释到24个COG类别中。其中,“一般功能基因”是最大类别,包含8 330 条unigene,占被注释到unigene总数的22.88%;其次是“蛋白质翻译后修饰与转运,分子伴侣”,包含4 128条unigene;而“核酸结构”是最小的类别,仅包含11条unigene。此外,有2 491条unigene参与了碳水化合物运输与代谢(图2)。
利用GO对获得辽东栎unigene进行功能分类,共有43 766条unigene被注释到生物学过程、细胞组分和分子功能3 个大类别中。其中,36 372条unigene归入生物学过程,18 876 条unigene归入细胞组分以及40 358条unigene归入分子功能。3个大的类别又被划分为45个小的类别(图3)。代谢过程是生物学过程中的最大类别,包含31 427条unigene;细胞是细胞组分中的最大类别,包含12 764条unigene;蛋白结合是分子功能中的最大类别,包含28 892条unigene。

图2 辽东栎unigene的COG分类
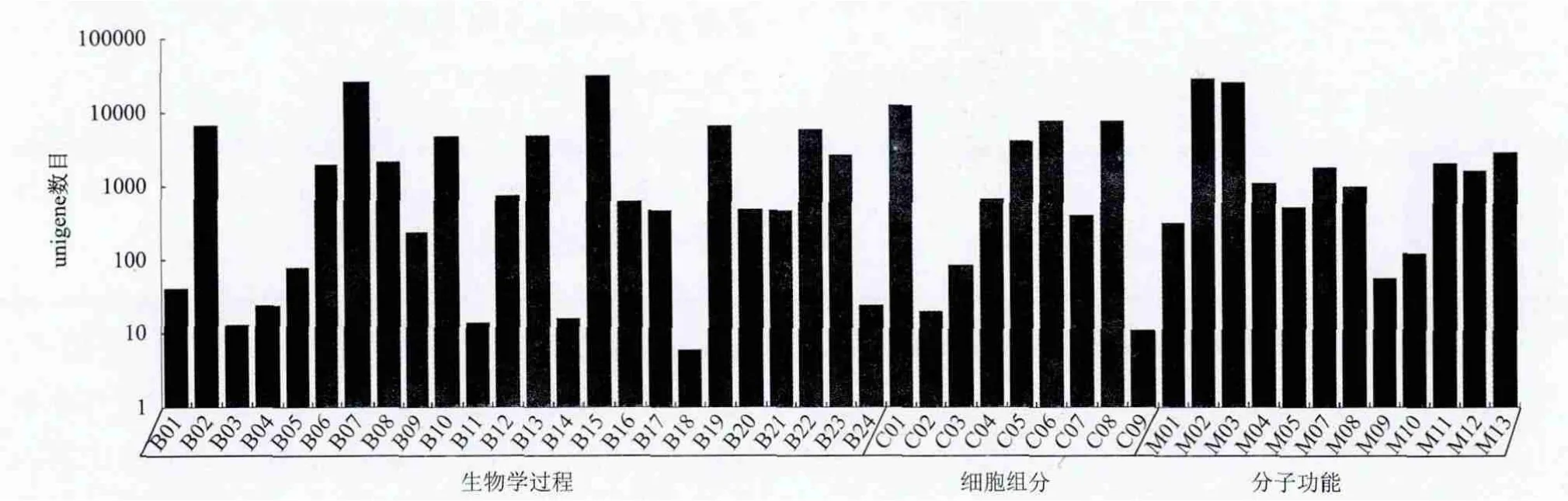
图3 辽东栎unigene的GO分类
2.4 代谢途径分析和淀粉合成基因的筛选
将辽东栎的unigene序列映射到KEGG数据库的参考代谢通路中,共有11 468条unigene参与到185个代谢通路中。其中包含unigene最多的是代谢通路是剪接体(ko03010),共有1 161条unigene。其次是内质网上的蛋白加工(ko04141),包含630条unigene。而参与淀粉与蔗糖的代谢通路(ko00500)的unigene共有434条(表1)。

表1 辽东栎中包含unigene数目最多的10个代谢通路
结合KEGG数据库中参考pathway的关于淀粉与蔗糖代谢通路中发掘到的unigene及在其他公共数据库中的注释,共统计筛选出67条参与淀粉合成的unigene,编码9个关键酶,其中5个unigene编码α-糖苷酶;17个unigene编码β-呋喃果糖苷酶;9个unigene编码己糖激酶;10个unigene编码果糖激酶;4个unigene编码葡萄糖-6-磷酸异构酶;4个unigene编码葡萄糖磷酸变位酶;8个unigene编码葡萄糖-1-磷酸腺苷酰基转移酶;6个unigene编码淀粉合成酶及4个unigene编码1,4-α-葡聚糖分支酶(表2)。

表2 辽东栎中参与淀粉合成的酶
2.5 SSR分析
利用MISA软件在辽东栎的13 380条unigene中共搜索到15 901个SSR位点,占unigene总序列数的13.97%,平均每1.28 kb出现1个SSR,其中包含有两个及两个以上SSR的unigene共有2 521条。二核苷酸和三核苷酸重复类型分别占SSR总数的60.07%和38.09%;四核苷酸重复类型占SSR总数的1.57%;而五核苷酸和六核苷酸重复类型在辽东栎中转录组序列中含量较少,仅占SSR总数的0.19%和0.09%。除此之外,不同核苷酸的重复次数也有很大的变化(表3)。
3 讨论
基于高通量测序技术的转录组学研究是一种非常高效、可靠的发掘功能基因手段,且在淀粉的合成及代谢中也已得到广泛应用[20,21]。目前,高通量测序技术主要是Roche公司的454测序技术、Illumina公司和ABI公司相继推出的Solexa和SOLid测序技术。其中,Solexa测序技术相对于其他两种技术在测序成本和数据量输出方面更具优势[22]。前人的研究表明,不同组织的混合取样,可在节约试验成本的基础上发掘到更多的转录本[23]。因此,本研究采用辽东栎的芽、花、叶和果实的混合样品进行转录组测序。栎属植物的基因组大小约为539-921 Mb[24],本研究共获得3.8 Gb的有效数据量,约覆盖辽东栎基因组的4.13-7.05倍。同时利用在对Illumina Solexa Hiseq 2000测序数据进行拼接过程中表现非常优异的Trinity软件[25]对本研究的数据进行拼接处理,共获得95 800条unigene,其中有38 163条unigene未在Blastx同源性搜索中得到注释,但大多数片段小于1 000 bp(37 752条,占98.92%),因此这些片段可能是由于较短而未与公共数据库中的序列比对上,也可能是短的非编码序列或者是新的基因。
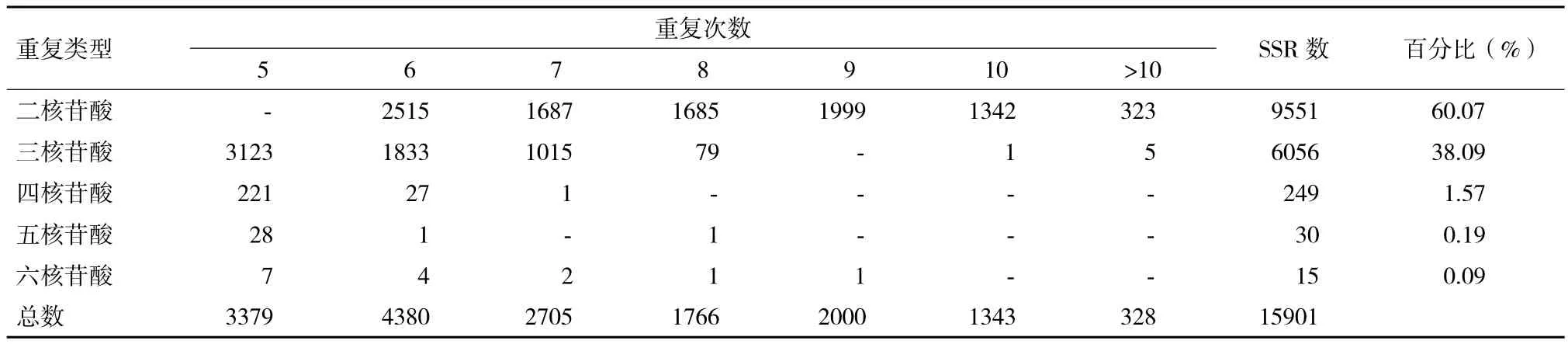
表3 辽东栎中的SSR位点的数量与分布
COG和GO的功能分类对初步了解基因的功能起着重要作用,而KEGG数据库中的参考pathway不仅可以推测基因的功能,而且可以研究基因在不同代谢通路中所在位置及作用,三者相辅相成,成为新物种中发掘功能基因的重要手段[19]。本研究通过KEGG 数据库中的pathway分析筛选与淀粉合成相关的基因,同时结合所筛选到的unigene在本研究中其他数据库中的功能注释,从而进一步确保了所获得的基因的可靠性。
鉴于分布广泛和多态性较高的特性,SSR标记已被广泛应用于分子标记辅助选择育种(Molecular marker-assisted selection,MAS)和利用分子标记通过关联分析(Association mapping)发掘与好的农艺性状连锁紧密的相关基因[26],尤其是对于多年生的植物,可大大缩短育种年限[27]。本研究在辽东栎中发掘到15 901个SSR位点,其中二核甘酸和三核苷酸的重复占到总数的98.16%,为了保证SSR位点的潜在多态性,我们在筛选过程中对于四、五和六核苷酸的最小重复次数同样设置为5,一定程度上影响到了这3类核苷酸重复在总SSR位点中所占比例。
本研究通过高通量的测序,获得了大量的辽东栎转录组序列,不仅丰富了辽东栎的基因库,而且为辽东栎及其他栎类淀粉合成基因的克隆与功能研究奠定了基础。发掘到的SSR位点可通过进一步的开发为辽东栎的进化与多样性分析及辽东栎的遗传图谱的构建和QTL(Quantitative Trait Loci)的定位提供了数据支持。
4 结论
本研究通过Solexa Hiseq 2000高通量测序,获得3.8 Gb的辽东栎转录组序列,拼接获得95 800条unigene,发掘出67条参与淀粉合成的unigene以及15 901个SSR位点。
[1] Balat M, Balat H. Recent trends in global production and utilization of bio-ethanol fuel [J] . Applied Energy, 2009, 86:2273-2282.
[2] Hahn-Hägerdal B, Galbe M, Gorwa-Grauslund MF, et al. Bioethanol-the fuel of tomorrow from the residues of today[J] . Trends in Biotechnology, 2006, 24(12):549-556.
[3] 谢光辉, 郭兴强, 王鑫, 等.能源作物资源现状与发展前景[J] .资源科学, 2007, 29(5):74-80.
[4] 吴创之, 周肇秋, 阴秀丽, 等.我国生物质能源发展现状与思考[J] .农业机械学报, 2009, 40(1):91-99.
[5] 李碧芳.发展生物质能源对能源安全和粮食安全的影响[J] .生态经济, 2010, 3:41-42.
[6] 方炎明, 张聪颖, 虞木奎, 等.栎树繁殖生物技术进展[J] .生物技术通报, 2011(4):60-65.
[7] 陈婧, 马履一, 段劼, 等.中国林业生物质能源发展现状[J] .林业实用技术, 2012, 11:16-19.
[8] 田玉峰, 李安平, 谢碧霞, 等.橡实淀粉生物乙醇化橡实品种和菌种的筛选[J] .食品科学, 2011, 32(7):207-210.
[9] 李安平, 田玉峰, 谢碧霞, 等.橡实淀粉生料酒精发酵与传统酒精发酵的能耗和成分组成比较[J] .江西农业大学学报, 2012,34(5):1032-1038.
[10] Soni PL, Sharma H, Dun D, et al. Physicochemical properties ofQuercus leucotrichophora(Oak)starch [J] . Starch/Stärke, 1993,45(4):127-130.
[11] Stevenson DG, Jane JL, Inglett GE. Physicochemical properties of pin oak(Quercus palustrisMuenchh.)acorn starch [J] . Starch/Stärke, 2006, 58(11):553-560.
[12] Derory J, Léger P, Garcia V, et al. Transcriptome analysis of bud burst in sessile oak(Quercus petraea)[J] . New Phytologist,2006, 170(4):723-738.
[13] Lesur I, Durand J, Sebastiani F, et al. A sample view of the pedunculate oak(Quercus robur)genome from the sequencing of hypomethylated and random genomic libraries [J] . Tree Genetics& Genomes, 2011, 7(6):1277-1285.
[14] Ueno S, Le Provost G, Léger V, et al. Bioinformatic analysis of ESTs collected by Sanger and pyrosequencing methods for a keystone forest tree species:oak [J] . BMC Genomics, 2010, 11:650.
[15] 刘红亮, 郑丽明, 刘青青, 等.非模式生物转录组研究[J] .遗传, 2013, 35(8):955-970.
[16] 罗伟祥, 郝怀晓, 薛安平.橡树资源-优质林木生物质能源发展战略研究[J] .生物质化学工程, 2006, 40(B12):147-152.
[17] Huang LL, Yang X, Sun P, et al. The first Illumina-based de novo transcriptome sequencing and analysis of safflower flowers [J] .PloS One, 2012, 7(6):e38653.
[18] Haas BJ, Papanicolaou A, Yassour M, et al.De novotranscript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis [J] . Nature Protocols, 2013,8(8):1494-1512.
[19] Wang R, Xu S, et al.De novosequence assembly and characterization ofLycoris aureatranscriptome using GS FLX Titanium platform of 454 pyrosequencing [J] . PloS One, 2013, 8(4):e60449.
[20] Tao X, Fang Y, Xiao Y, et al. Comparative transcriptome analysis to investigate the high starch accumulation of duckweed(Landoltia punctata)under nutrient starvation [J] . Biotechnology for Biofuels, 2013, 6(1):72.
[21] Kaminski KP, Petersen AH, Sønderkær M, et al. Transcriptome analysis suggests that starch synthesis may proceed via multiple metabolic routes in high yielding potato cultivars [J] . PloS One,2012, 7(12):e51248.
[22] Shu S, Chen B, et al.De novosequencing and transcriptome analysis ofWolfiporia cocosto reveal genes related to biosynthesis of triterpenoids [J] . PloS One, 2013, 8(8):e71350.
[23] Zhou Y, Gao F, Liu R, et al.De novosequencing and analysis of root transcriptome using 454 pyrosequencing to discover putative genes associated with drought tolerance inAmmopiptanthus mongolicus[J] . BMC Genomics, 2012, 13:266.
[24] Kole, Chittaranjan. Fagaceae Trees. //In genome mapping and molecular breeding in plants[M] . Springer, 2007, 7:161-184.
[25] Li SW, Yang H, Liu YF, et al. Transcriptome and gene expression analysis of the rice leaf folder,Cnaphalocrosis medinalis[J] . PloS One, 2012, 7(11):e47401.
[26] O’Malley DM, McKeand SE. Marker assisted selection for breeding value in forest trees [J] . Forest Genetics, 1994, 1:207-218.
[27] Neale DB, Kremer A. Forest tree genomics:growing resources and applications [J] . Nature Reviews Genetics, 2011, 12:111-122.
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
农业技术与装备(2022年6期)2022-08-17
肝博士(2022年3期)2022-06-30
辽宁省博物馆馆刊(2020年0期)2020-08-13
东坡赤壁诗词(2019年3期)2019-07-05
西安工程大学学报(2016年6期)2017-01-15
中国边疆民族研究(2016年0期)2016-12-18
中国粮油学报(2016年1期)2016-02-06
食品工业科技(2014年23期)2014-03-11
